Pedestal Formation by Pathogenic Escherichia coli: A Model System for Studying Signal Transduction to the Actin Cytoskeleton
Dynamic rearrangement of the actin cytoskeleton in response to signal transduction plays a fundamental role in the regulation of cellular functions. Understanding actin dynamics therefore represents one of the challenges of modern cell biology. Several pathogens have evolved diverse strategies to trigger rearrangement of the host actin cytoskeleton to facilitate and enhance their infection processes. By manipulating the actin assembly machinery or signalling routes leading to its activation, pathogens can either block or induce phagocytosis, drive intracellular motility, and exploit their host cells in other ways. Analyses of host pathogen interactions have not only broadened our knowledge of how these pathogens cause disease, but have also emerged as model systems in the study of cellular actin dynamics (for examples, see Frischknecht and Way, 2001). The focus of this article is on the formation of cytoskeletal rearrangements called actin pedestals induced by the diarrhoeagenic extracellular bacterial pathogens enteropathogenic Escherichia coli (EPEC) and enterohaemorrhagic E. coli (EHEC) as systems to study signalling and actin assembly at the plasma membrane.
Importantly, the histopathological changes associated with the A/E phenotype in vivo can be mimicked in cell culture [(Knutton et al., 1987), see Fig. 1], which allows to define the molecular mechanisms employed by EPEC and EHEC to induce cytoskeletal rearrangements. The ability to form actin pedestals on cultured cells furthermore correlates with the ability of EPEC and EHEC to colonize the intestine and cause disease in human and other animal hosts (e.g., Donnenberg et al., 1993).
 |
| FIGURE 1 Actin pedestal formation induced by EHEC and EPEC in vivo and on the surface of cultured cells. (A) Transmission electron micrograph showing the characteristic attaching and effacing (A/E) lesion formation of the enterohaemorrhagic Escherichia coli (EHEC) O157:H7 strain 86/24 observed in piglet colon. Note the intimate attachment, localized loss of microvilli, and formation of a raised, pedestal-like structure beneath the bacterium that characterizes this lesion (courtesy of Florian Gunzer, Institute of Medical Microbiology, Hannover Medical School, Germany). (B) Scanning electron micrograph of enteropathogenic E. coli (EPEC) O127:H6 strain E2348/69 (pseudocoloured in red) sitting on top of pedestals induced on the surface of cultured routine embryonic fibroblast cells upon infection that resemble A/E lesions formed by EPEC in vivo. (C) EHEC O157:H7 strain 86/24-induced actin pedestal formation as visualized by fluorescence actin staining using AlexaFluor 594 phalloidin to specifically label F-actin (shown in red). EHEC bacteria shown in green were detected with monoclonal antibodies against EspE (Deibel et al., 1998) and AlexaFluor 488- conjugated goat antimouse secondary antibodies. Bars: 1 µm. |
Both pathogens translocate their own receptor, the translocated intimin receptor (Tir, EspE), which binds to the bacterial surface protein intimin, via a type III secretion system into the underlying host cell. EPEC Tir becomes tyrosine phosphorylated upon insertion into the host cell membrane and binding to intimin. This phosphorylation is critical for actin pedestal formation by EPEC (Kenny, 1999), as it allows recruitment of the cellular signalling adaptor protein Nck to bacterial attachment sites. Nck, in turn, triggers by a so far unknown mechanism recruitment and activation of NWASP. Both Nck and N-WASP are essential host proteins for pedestal formation induced by EPEC, as cells lacking either Nck or N-WASP are resistant to actin pedestal formation by EPEC (Gruenheid et al., 2001; Lommel et al., 2001) (for N-WASP, see Fig. 2). N-WASP is a member of the WASP/Scar family of cellular nucleation-promoting factors and has emerged as a central node protein that regulates actin polymerisation by activating the Arp2/3 complex, a main factor for nucleation of actin filaments in response to multiple upstream signals.
In contrast, EHEC Tir is not tyrosine phosphorylated and pedestals are formed independently of Nck. Despite that, EHEC-induced pedestal formation still depends on N-WASP function (Lommel et al., 2004) to promote Arp2/3 complex-mediated actin polymerisation. Thus, EPEC and EHEC have evolved different strategies to trigger cellular signalling routes leading to actin assembly, which converge in recruitment and activation of N-WASP to promote Arp2/3-mediated actin polymerisation.
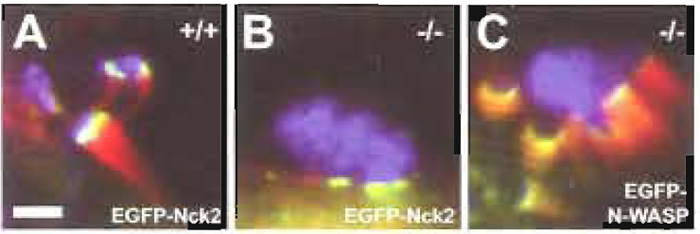 |
| FIGURE 2 EGFP-tagging and knockout cell lines as tools for analysis of the molecular mechanism of actin pedestal formation induced by EPEC. Cultured routine embryonic fibroblasts expressing (+/+, A) or lacking N-WASP (-/-, B and C) were infected with EPEC and examined by immunofluorescence microscopy. F-actin is shown in red as detected by fluorescence actin staining with Alexa- Fluor 594 phalloidin, bacteria are shown in blue as detected with anti-EspE monoclonal antibodies in combination with AlexaFluor 350-conjugated goat antimouse secondary antibodies, and the host proteins Nck2 (A and B) and N-WASP (C) expressed ectopically with an EGFP tag are shown in green. Whereas EPEC induced the formation of prominent pedestals in N-WASP-expressing cells (+/+, A), they were unable to trigger actin accumulation on N-WASPdefective cells (-/-, B). The bacterial ability to direct actin reorganisation in N-WASP-defective fibroblasts was restored by providing EGFP-tagged N-WASP by transient transfection (C). Together, this clearly demonstrates that N-WASP is a host cell protein essential for pedestal formation induced by EPEC. Recruitment of the host cell signalling adaptor protein Nck2 is triggered by EPEC independently of actin accumulation, as was revealed by ectopic expression of EGFP-tagged Nck2 in either N-WASP-expressing (+/+, A) or NWASP- defective cells (-/-, B). Bar: 2µm, (valid for A-C). |
A Cell Lines, Bacterial Pathogen Strains, and Culture Media Reagents
1. Cells
Epithelial cell lines used commonly for EPEC and EHEC infection experiments are HeLa and HEp-2 cells, as well as the intestinal epithelial cell lines T84 or Caco-2 (available from American Type Culture Collection). EPEC and EHEC will also adhere and induce the formation of actin pedestals on mouse embryonic fibroblast cell lines (MEFs), which are used routinely in our laboratory. Embryonic fibroblast cell lines offer the advantage that such cell lines can be established quite easily from conditional or conventional knockout mice, thus allowing the analysis of specific host proteins in actin pedestal formation induced by EPEC and EHEC.
2. Bacterial Pathogen Strains
Prototype enteropathogenic and enterohaemorrhagic E. coli strains used in analyses of the molecular mechanism of actin pedestal formation are enteropathogenic E. coli strain E2348/69 (O127:H6) (Levine et al., 1978), enterohaemorrhagic E. coli strain 86/24 (O157:H7) [isolated from an outbreak in Walla Walla, WA., U.S.A. (Griffin et al., 1988)], and enterohaemorrhagic E. coli strain EDL933 (O157:H7) (American Type Culture Collection #700927; for genome sequence information, see Perna et al., 2001).
Dulbecco's modified eagle medium (DMEM), low glucose (Invitrogen Corp., GIBCO #31885-023), fetal bovine serum (Sigma-Aldrich #F 7524), L-glutamine (Invitrogen Corp., GIBCO #25030-024), penicillin/ streptomycin (Invitrogen Corp., GIBCO #15070-063), Luria-Bertani (LB) broth agar (e.g., BD #244520), LB broth (e.g., BD #244620), and HEPES (Sigma-Aldrich #H 3375), fibronectin (pure) (Roche #1051407).
B. Constructs and Reagents for Expression of GFP-Tagged Host Proteins
- Transfection reagent, e.g., FuGENE 6 (Roche #1 814 443)
- A set of vectors for construction of EGFP fusions is available from Clontech (BD Biosciences).
- EGFP-N-WASP: as described (Lommel et al., 2001)
- EGFP-Nck2: as described (Scaplehorn et al., 2002)
C. Additionally Needed Reagents and Plasticware
1. Basics
General equipment and plasticware for molecular biology and cell culture techniques.
Twenty-four-well cell culture plates (e.g., Corning Inc. #3524), 12-mm round glass coverslips (e.g., Assistent #1001, thickness 0.17mm), absolute ethanol (Sigma-Aldrich #E 7023), HCl (37%) (Sigma-Aldrich #H 7020), lint-free absorptive paper [e.g., GB002, Schleicher& Schuell #10427736 (58 × 68cm) and #10485285 (22.2 × 22.2cm)], large square plastic dish (e.g., 24.5 × 24.5-cm polystyrene dish with lid, Sigma-Aldrich #Z37,165-3), Parafilm M (e.g., Fisher Scientific #917 00 02), forceps with curved fine tips (e.g., coverslip forceps Dumont #11251-33), NaCl (Sigma-Aldrich #S 7653), KCl (Sigma-Aldrich #P 1338), Na2HPO4 (Sigma- Aldrich #S 7907), KH2PO4 (Sigma-Aldrich #P 0662), paraformaldehyde (PFA), (Sigma-Aldrich #P 6148), NaOH (Merck/VWR International #109913), Triton X- 100 (Sigma-Aldrich #T 8532), normal goat serum (Invitrogen Inc.: GIBCO #16210-064), bovine serum albumin (BSA, Sigma-Aldrich #A 2153), goat antimouse Alexa Fluor 350- or Alexa Fluor 488-conjugated secondary antibodies (Molecular Probes #A-11045 and #A-11001), DAPI (e.g., Molecular Probes, #D-1306), fluorophore- coupled phalloidin [e.g., Alexa Fluor 594 phalloidin (red fluorescence, Molecular Probes #A- 12381)], glycerol (87%, analytical grade) (e.g., Merck/VWR International #104094), Mowiol 4-88 (Calbiochem #475904), Tris base (e.g., Trizma base, Sigma-Aldrich #T 1503), n-propyl gallate (SigmaAldrich #P 3130), and SuperFrost microscope glass slides (Fisher Scientific #9161161).
1. Centrifuges
Tabletop centrifuge (e.g., centrifuge 5414D, Eppendorf #5425 000.219), centrifuge equipped with rotor suitable for microtiter plates, and 15- and 50-ml polypropylene tubes (e.g., centrifuge 5810, Eppendorf #5810 000.017 with rotor A-4-81 and A-4-81-MTP).
2. Clean Benches and Cell Incubators
Clean bench and cell culture incubator suitable for work with EPEC and EHEC pathogens in accordance with respective national safety regulations.
3. Microscope
Inverted microscope (e.g., Axiovert 135TV, Carl Zeiss Jena GmbH) equipped for epifluorescence microscopy with 40×/1.3NA and 100×/1.3 NA Plan-NEOFLUAR oil immersion objectives, 1.6 and 2.5 optovar magnification, electronic shutters (e.g., Uniblitz Electronic 35-mm shutter including driver Model VMMD-1, BFI Optilas) to allow for computercontrolled opening of the light paths, excitation and emission filters (Omega Optical Inc. or Chroma Technology Corp.) to enable three-colour epifluorescence, and mercury short arc lamp (Osram, HBO103W/2) for fluorescence light path.
4. Data Acquisition
Preferably a back-illuminated, cooled chargecoupled device (CCD) camera (e.g., Princeton Research Instruments TKB 1000x800, SN J019820; Controller SN J0198609) driven, for instance, by IPLab (Scanalytics Inc.) or Metamorph software (Universal Imaging Corporation).
Solutions
- Cell culture growth media: Cell culture growth medium suitable for the propagation of the cell line chosen. We use DMEM, low glucose supplemented with 10% heat-inactivated fetal bovine serum, 2 mM L-glutamine, and 50 U/ml each of penicillin and streptomycin for propagation of our embryonic fibroblast cell lines.
- Cell culture growth media for bacterial infection experiments: For bacterial infection experiments, omit penicillin and streptomycin from growth media starting a day prior to infection.
- Standard bacterial cultures: Prepare LB broth and LB agar plates according to standard protocols (e.g., Maniatis et al., 1982).
- Preactivating culture of EPEC: DMEM, low glucose for culturing EPEC prior to infection of cell monolayers.
- Preactivating culture of EHEC: DMEM, low glucose, supplemented with 100mM HEPES, pH 7.4, for culturing EHEC prior to infection of cell monolayers.
- Phosphate-buffered saline (PBS): 140mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, pH 7.4
- Fixative: Prepare a 4% solution of PFA in PBS, pH 7.4. Paraformaldehyde is very toxic, work in fumehood when preparing stock, do not inhale, and wear gloves. To prepare 100ml, add 4g PFA to 90ml PBS. In order for the PFA to dissolve, heat the solution to 60-65°C with continuous stirring. If necessary, adjust the pH to 7.4 by adding NaOH (be patient!). Do not heat solution above 70°C, as PFA will degrade. Let cool to room temperature, check the pH, and adjust with PBS to full volume. Filter through paper filter to remove insoluble aggregates and store in aliquots (e.g., 15 and 50ml) at -20°C.
- Cell permeabilization: Fixative supplemented with 0.1% Triton X-100 just prior to use. Make up a 10% stock solution of Triton X-100 in PBS and store at 4°C.
- Antibody diluent: 1% BSA in PBS. Prepare and store in suitable aliquots (1-2ml) at -20°C.
- Blocking solution: 10% normal goat serum in PBS. Prepare from frozen aliquots of serum just prior to use.
- Antibody Mixtures: Dilute antibodies just prior to use to appropriate working concentration in 1% BSA in PBS. The ideal concentration will result in a strong signal with no or little background staining and has to be established experimentally for each new antibody. To start, follow instructions given by the supplier. When using a concentrated primary antibody, a 1:100 dilution resulting in about 10µg/ml should be a good starting point for immunofluorescence microscopy. Secondary antibodies conjugated to fluorophores are available from numerous suppliers. In our hands, secondary reagents coupled to Alexa Fluor dyes from Molecular Probes have worked very well. Note, however, that most polyclonal antisera will exhibit unspecific cross-reaction with EPEC and EHEC bacteria. A way to avoid problems associated with unspecific labelling of bacteria is to use monoclonal reagents (see general comment in Section IV). We store our antibodies in the dark at 4°C in a refrigerator. For longterm storage, antibodies may be stored at -20 or -80°C. Follow the recommendations given by the supplier.
- Mounting medium: Weigh out 6g glycerol and add 2.4 g Mowiol 4-88. Stir thoroughly. Add 6 ml aqua dest and mix for several hours at room temperature. Add 0.2ml 0.2M Tris-Cl, pH 8.5, and heat to 60°C for 10 min. Remove insoluble material by centrifugation at 6000g for 30min. Store in aliquots at -20°C. In order to reduce photobleaching during fluorescence microscopy, add 2.5-5mg/ml n-propyl gallate prior to use (Giloh and Sedat, 1982).
EPEC and EHEC pose a significant threat to human health, especially EHEC with its low infectious dose of ~10-100cfu. When working with EPEC and EHEC, always wear protective clothing and work under an appropriate clean bench in accordance with national safety regulations. Decontaminate all materials that have been in contact with the pathogen.
Steps
- Pretreat 12-mm round glass coverslips as follows: wash coverslips in a mixture of 60% absolute ethanol and 40% concentrated HCl for 30 min and rinse extensively with aqua dest (heating in a microwave oven is helpful). To dry coverslips, spread on lint-free absorptive paper. Sterilize for tissue culture either by autoclaving on the dry cycle at 220°C or by exposing to ultraviolet light for 45 min in a culture dish.
- On day 1, streak EPEC and EHEC from frozen glycerol stocks onto fresh LB agar plates and incubate overnight at 37°C. Keep plates in the refrigerator until liquid overnight cultures are started.
- On day 2, seed cells onto 12-mm pretreated coverslips in a 24-well tissue culture plate in cell culture growth media without antibiotics. Incubate overnight in a tissue culture incubator supplemented with CO2 to allow the cells to adhere to the coverslips. For MEFs, we find microscopic analysis of actin pedestal formation easiest if cells have reached about 80% confluency at the time of analysis; the number of cells seeded should be adjusted accordingly. To reduce detachment of MEFs infected with EHEC, use fibronectin coated coverslips.
- Start an overnight liquid culture of EPEC or EHEC in 5ml LB medium with bacteria from the streaked plate. In our experience, small inoculation loads result in a higher infection efficiency. Grow overnight with aeration at 37°C on a rotary shaker at 180rpm.
- On day 3, collect bacteria from the 0.5-ml overnight culture by centrifugation for 3min at 4500 rpm in a tabletop centrifuge. Wash bacterial pellet twice in DMEM and inoculate 25 ml DMEM (EPEC) or 25 ml DMEM supplemented with 100mM HEPES, pH 7.4 (EHEC). Grow at 37°C at 180rpm for 3 h.
Environmental cues such as temperature, pH, and osmolarity, as well as growth phase, have been described to influence the transcriptional regulation of expression of virulence factors, e.g., of the type III secretion system. Media conditions that appear to stimulate virulence are those considered to mimic the gastrointestinal tract (e.g., Beltrametti et al., 1999; Kenny et al., 1997). In our experience, preactivation of EPEC and EHEC by growth for 3 h in DMEM for EPEC or DMEM supplemented with 100 mM HEPES, pH 7.4, for EHEC is sufficient for reproducible efficient bacterial infection of cultured cell monolayers. In our hands, the morphological appearance of preactivated bacterial cultures correlates with their infectious capability: After 3h, EPEC should be found aggregated to small clumps of bacteria but should not grow in long rows. Aggregation likely reflects the expression of type IV bundle-forming pili, rope-like appendages that represent an important additional virulence factor found to enhance initial adherence and virulence of EPEC (Bieber et al., 1998). EHEC, which lack bundleforming pili, should be present as single bacteria in preactivation cultures, but again should not grow in long rows.
- In the meantime, exchange cell culture medium of cell monolayers with 1 ml per well of fresh cell culture medium without antibiotics.
- Add 10µl of the bacterial 3-h preactivation culture into each well. Initiate infection by brief centrifugation for 5 min at 650 g in a centrifuge prewarmed to at least room temperature, allowing bacteria and cells to make contact with each other.
- Place in an incubator supplemented with 7.5% CO2 at 37°C for 1h. Incubation at lower temperatures results in a reduced infection efficiency.
- Remove media containing nonattached bacteria by aspiration and gently replace with 1 ml of fresh prewarmed culture medium without antibiotics per well. Be careful not to let the cells dry out when exchanging the medium.
- Repeat medium changes every hour until a total infection time of 4.5 to 5 h has been reached.
In our hands, longer infection times generally result in pedestals of increased length, facilitating analysis of pedestal composition. In addition, after short incubation times, EPEC bacteria are found mostly adhered in aggregates, which may confound analysis. Later during infection, EPEC bacteria will distribute from aggregates, making it easier to distinguish single pedestals. However, too long incubation results in increased cell death and detachment of cells, especially when working with EHEC.
- After the incubation period, remove the medium by aspiration and gently wash the coverslips twice with PBS prewarmed to 37°C to remove unattached bacteria.
- For fixation, immerse each coverslip with 500µl of fixative prewarmed to 37°C and incubate at room temperature for 20min.
- After fixation, gently wash twice with PBS and store in PBS at 4°C until proceeding to cell permeabilization and immunostaining.
B. Alternative Protocol: EPEC and EHEC Infections of Transiently Transfected Cell Monolayers
For experiments involving ectopic expression of host cell molecules, e.g., tagged with GFP, designed to analyse their role in actin pedestal formation induced by EPEC or EHEC (see general comments, Section IV), follow the basic protocol with the exceptions that cells are already seeded on day 1, and day 2 will be required for transfection of host cell monolayers with plasmid DNA in addition to starting bacterial overnight cultures.
Steps
- On day 1, seed cells in regular cell culture growth media on coverslips as described in the basic protocol with the modification that the number of cells seeded should be reduced to allow for the additional day needed for transfection. Again, about 80% confluency of MEFs should be reached at the time of bacterial infection.
- On day 2, prior to transfection, exchange the cell culture growth medium for 0.5ml growth medium without antibiotics. Transfect cells growing on coverslips in 24-well plate 12-24h overnight according to standard protocols, e.g., using FuGENE 6 transfection reagent. We use 0.2µg of DNA per well of a 24-well plate with a ratio of FuGENE 6 to DNA of 3:1. In our hands, plasmid DNA prepared with MaxiPrep kits, e.g., from Qiagen or Invitrogen Corp. is sufficiently pure for cell transfection.
C. Immunofluorescence Microscopy
See general comments about immunofluorescence microscopy analysis of EPEC- and EHEC-infected cells.
Steps
- Permeablize cells in 0.1% Triton X-100 in PBS for 1 min.
- Gently wash twice with PBS.
- Prepare a humid chamber: Lay out a large square plastic dish (e.g., 24.5 × 24.5-cm polystyrene dish with lid) with a moistened piece of absorptive filter paper and coat with a layer of Parafilm M.
Perform all blocking and immunolabelling steps as follows: Using forceps, place coverslips with the cell side facing downward onto a drop of about 20µl of blocking or antibody mixture spotted onto parafilm in the humid chamber. This helps minimize the amount of antibody needed without letting the cells dry out. In order to minimize the loss of cells when moving the coverslips after each incubation step, pipette a small amount of PBS right next to the edge of each coverslip after each incubation step. This will cause the coverslips to float on top of the PBS and will allow easy removal of the coverslips using forceps. Wash coverslips by carefully submerging them successively into three small beakers filled with PBS. In between individual washing steps, drain residual PBS by carefully streaking the edge of the coverslips across thin absorptive filter paper without allowing the cells to dry out.
- Block with 10% normal goat serum in PBS for 20 min in humid chamber.
- Stain with primary antibody mixture for 1 h at room temperature in humid chamber.
- Wash three times with PBS. For polyclonal primary antibodies, include 0.1% Triton-X100 during first two washing steps.
- Stain with secondary antibody mixture for 1h at room temperature in humid chamber in the dark. To specifically label F-actin, use fluorescent dye-coupled phalloidin, e.g., Alexa Fluor 594 phalloidin (red fluorescence), added at a concentration of 1-2.5U/ml. To detect Leatuis by staining bacterial DNA, blue fluorescent DAPI may be added.
- Wash three times with PBS. For polyclonal primary antibodies, include 0.1% Triton-X100 during first two washing steps.
- To mount coverslips onto microscope glass slides, place drops of about 5µl mounting medium on ethanol-wiped slides. Avoid air bubbles. Using forceps, gently place coverslips on mounting medium with the cell side facing downward. Once in contact with the mounting medium, avoid moving the coverslips as this will cause cells to be ripped off. Carefully remove access mounting media or residual PBS by placing a piece of absorptive filter paper onto the glass slides without moving the coverslips.
- Dry mount coverslips for 3-4h at room temperature in the dark or overnight prior to microscopic observation when using oil immersion lenses.
- Store samples in the dark at 4°C. It is advisable to complete photography of samples within 1-2 weeks, as background fluorescence increases over time.
- Using epifluorescence and appropriate emission filters, infected cells are detected readily by fluorescence actin staining (FAS) of actin pedestals. Search for interesting cells (e.g., both transfected and infected) using low magnification (e.g., 40x). To resolve subcellular structures such as pedestals, switch to higher magnification (100x) with or without optovar magnification. Always use minimal exposure time required to get a good signal-to-noise ratio.
It is important to consider that polyclonal antibodies are likely to exhibit cross-reactivity against E. coli, which will lead to unspecific labeling of EPEC or EHEC bacteria and may confound the analysis of the role of a specific protein in actin pedestal formation by immunofluorescence microscopy. Thus, it may be difficult to discern between recruitment of a protein to the very tips of pedestals or bacterial attachment sites and unspecific labeling of bacteria, especially when using low magnification. Always test secondary reagents for unspecific labeling of bacteria. Consider using affinity-purified reagents. For polyclonal primary reagents, try addition of 0.1% Triton-X100 to PBS during washing steps. Cross-reaction of polyclonal reagents is no problem when immunolabelling is only performed to detect bacteria, as is the case in the examples shown in Fig. 2, in which EPEC bacteria were detected with monoclonal antibodies against bacterial EspE (Deibel et al., 1998) and Alexa Fluor 350 fluorescent-labelled goat antimouse secondary antibodies, whereas F-actin was stained with fluorescent Alexa Fluor 594 phalloidin, and the host cell protein Nck was visualized with the help of an EGFP tag. There are reports in the literature about preabsorption of antibodies against fixed bacteria prior to immunolabelling, which when tried in our laboratory was of little success. Another possible solution to overcome the problem may be the use of monoclonal secondary reagents (e.g., rat antimouse), which are available from various suppliers (e.g., Zymed Laboratories), although so far not as conjugates with AlexaFluor dyes.
Acknowledgments
We thank Florian Gunzer for the transmission electron micrograph of EHEC-infected piglet colon for Fig. 1, Klemens Rottner and Theresia Stradal for support and helpful discussions, and Brigitte Denker for excellent technical assistance. J.W. was supported by the DFG (SFB 621) and the Fonds der Chemischen Industrie.
References
Beltrametti, F., Kresse, A. U., and Guzman, C. A. (1999). Transcriptional regulation of the esp genes of enterohemorrhagic Escherichia coli. J. Bacteriol. 181, 3409-3418.
Campellone, K. G., and Leong, J. M. (2003). Tails of two Tirs: Actin pedestal formation by enteropathogenic E. coli and enterohemorrhagic E. coli O157:H7. Curr. Opin. Microbiol. 6, 82-90.
Deibel, C., Kramer, S., Chakraborty, T., and Ebel, F. (1998). EspE, a novel secreted protein of attaching and effacing bacteria, is directly translocated into infected host cells, where it appears as a tyrosine-phosphorylated 90kDa protein. Mol. Microbiol. 28, 463-474.
Donnenberg, M. S., Tzipori, S., McKee, M. L., O'Brien, A. D., Alroy, J., and Kaper, J. B. (1993). The role of the eae gene of enterohemorrhagic Escherichia coli in intimate attachment in vitro and in a porcine model. J. Clin. Invest. 92, 1418-1424.
Frischknecht, F., and Way, M. (2001). Surfing pathogens and the lessons learned for actin polymerization. Trends Cell Biol. 11, 30-38.
Giloh, H., and Sedat, J. W. (1982). Fluorescence microscopy: Reduced photobleaching of rhodamine and fluorescein protein conjugates by n-propyl gallate. Science 217, 1252-1255.
Gruenheid, S., DeVinney, R., Bladt, F., Goosney, D., Gelkop, S., Gish, G. D., Pawson, T., and Finlay, B. B. (2001). Enteropathogenic E. coli Tir binds Nck to initiate actin pedestal formation in host cells. Nature Cell Biol. 3, 856-859.
Kenny, B. (1999). Phosphorylation of tyrosine 474 of the enteropathogenic Escherichia coli (EPEC) Tir receptor molecule is essential for actin nucleating activity and is preceded by additional host modifications. Mol. Microbiot. 31, 1229-1241.
Kenny, B., Abe, A., Stein, M., and Finlay, B. (1997). Enteropathogenic Escherichia coli protein secretion is induced in response to conditions similar to those in the gastrointestinal tract. Infect. Immun. 65, 2606-2612.
Knutton, S., Lloyd, D. R., and McNeish, A. S. (1987). Adhesion of enteropathogenic Escherichia coli to human intestinal enterocytes and cultured human intestinal mucosa. Infect. Immun. 55, 69- 77.
Levine, M. M., Bergquist, E. J., Nalin, D. R., Waterman, D. H., Hornick, R. B., Young, C. R., and Sotman, S. (1978). Escherichia coli strains that cause diarrhoea but do not produce heat-labile or heat-stable enterotoxins and are non-invasive. Lancet 1, 1119-1122.
Lommel, S., Benesch, S., Rottner, K., Franz, T., Wehland, J., and Kuhn, R. (2001). Actin pedestal formation by enteropathogenic Escherichia coli and intracellular motility of Shigella flexneri are abolished in N-WASP-defective cells. EMBO Rep. 2, 850-857.
Maniatis, T., Fritsch, E. F., and Sambrook, J. (1982). "Molecular Cloning: A Laboratory Manual." Cold Spring Harbor Laboratory, Cold Spring Harbor, NY.
Nataro, J. P., and Kaper, J. B. (1998). Diarrheagenic Escherichia coli. Clin. Microbiol. Rev. 11, 142-201.
Perna, N. T., Plunkett, G., 3rd, Burland, V., Mau, B., Glasner, J. D., Rose, D. J., Mayhew, G. F., Evans, P. S., Gregor, J., Kirkpatrick, H. A., Posfai, G., Hackett, J., Klink, S., Boutin, A., Shao, Y., Miller, L., Grotbeck, E. J., Davis, N. W., Lim, A., Dimalanta, E. T., Potamousis, K. D., Apodaca, J., Anantharaman, T. S., Lin, J., Yen, G., Schwartz, D. C., Welch, R. A., and Blattner, F. R. (2001). Genome sequence of enterohaemorrhagic Escherichia coli O157:H7. Nature 409, 529-533.
Scaplehorn, N., Holmstrom, A., Moreau, V., Frischknecht, F., Reckmann, I., and Way, M. (2002). Grb2 and Nck act cooperatively to promote actin-based motility of vaccinia virus. Curr. Biol. 12, 740-745.
Support our developers




