Purification of Non-muscle Actin
In the past decade or so, a number of new actin regulatory proteins and their functions in the motility of nonmuscle cells have been unveiled. We now understand in some detail how extracellular stimuli are transmitted to the microfilament system via activation of the phosphatidylinositol cycle, rho-family of GTPases, causing polymerization of actin through the recruitment of WASp, WAVE, ARP2/3, or formin proteins. The polymerization of actin requires constant supply of actin kept in an unpolymerized form by proteins such as thymosin, cofilin, and profilin. Filamentcapping, cross-linking, destabilising, and sequestering proteins cooperate with monomeric (G)-actin to contribute to the dynamics of the system. Thus, complex machineries for the controlled polymerisation of actin are now being revealed (see Ridley et al., 2003; Innocenti et al., 2004).
Isoforms of actin differ in their affinities for their ligands, their functioning, and subcellular locations. Therefore, to understand the function of actin in nonmuscle cells, it is important to work with nonmuscle actin when studying actin-binding proteins (ABPs) from nonmuscle tissues. Unfortunately, this is often neglected because of the ready access to α-actin from rabbit skeletal muscle (Pardee and Spudich, 1982). A further complication is the covalent modification introduced in α-actin during the commonly used purification protocol involving acetone extraction of muscle tissue (Selden et al., 2000). This modification leads to the formation of stable dimers, explaining fast polymerisation kinetics and other properties thought to be characteristic for α-actin. However, i- and -actin are prepared easily from various nonmuscle tissues using the method described in this article.
II. MATERIALS AND INSTRUMENTATION
All chemicals (reagent grade) are from ICN or Sigma. Hydroxyl apatite is hypatite C of even lot number from Clarkson Chemical Co. (Williamsport, PA). This is the only hydroxyl apatite material we have used successfully to separate actin isoforms. DNase I is from ICN (Cat. No. 100579) and poly-(L-proline) is from Sigma (Cat. No. P-2129). CNBr-activated Sepharose 4B is from Amersham Biosciences (Cat. No. 17-0430). Empty columns are XK-16/20, XK-26/20, and XK-50/20 from Amersham Biosciences. All other chromatography equipments and matrices are from Amersham Biosciences.
Yeast nitrogen base without amino acids is from Difco (cat. No. 291940), adenine sulfate is from ICN (Cat. No. 100195), and the casamino acid vitamin assay is from Difco (Cat. No. 228820). All other media components are from Difco. Lysis of yeast cells is achieved with a purpose-designed bead mill (Innomed Konsult) using glass beads 0.25-0.5 mm in diameter (Mahlkörper MK2GX, Willy Bachofen). However, alternative procedures to break up yeast cells have been described (Jazwinski, 1990). Low-speed centrifugations are carried out in a Beckman-Coulter Avanti J-20XP with polypropylene tubes (50 ml; Beckman Cat. No. 357007) in a Beckman JA-25.0 rotor. Large-volume centrifugations are performed with polycarbonate centrifuge bottles (500ml; Beckman no. 335605) in a Beckman JLA-16.250 rotor. Preparative ultracentrifugations are done in a Beckman-Coulter L-series centrifuge with polycarbonate bottles (70ml; Beckman No. 355622) in Rotor Beckman 45Ti.
A. Preparation of Profilin: Actin Complexes from Bovine Thymus or Spleen
This protocol is used for the purification of profilin in complex with a mixture of the cytoplasmic β- and γ- actin isoforms. The isolation procedure involves affinity binding to poly-(L-proline) and chromatography on hydroxyl apatite to separate actin isoforms, either alone or in complex with profilin (Segura and Lindberg, 1984; Lindberg, 1988). The yield of profilin: actin complex is approximately 400 mg/kg of tissue. The following section describes a preparation starting with 700 g of calf thymus. All buffer volumes and column sizes refer to that preparation size. If calf thymus cannot be obtained, calf spleen can be used instead. Actin isoform distribution is approximately 60% β- actin and 40% γ-actin in thymus, whereas spleen contains slightly more β-actin. Spleen is easier to handle, but contains more proteolytic enzymes, which are difficult to control.
1. Affinity Purification of Profilin :Actin Complexes
- Homogenisation buffer (2 liters): 10 mM Tris-HCl, pH 7.6, 0.1M KCl, 0.1M glycine, 1% Triton X-100, and 0.5 mM, dithiothreitol (DTT)
- 2× PLP buffer (3 liters): 20mM Tris-HCl, pH 7.6, 0.2 M KCl, and 0.2 M glycine
- PLP elution buffer (500ml): make immediately before use. Mix 250ml of 2x PLP buffer with 150ml of dimethyl sulfoxide (DMSO) and 100ml distilled H2O. Mixing DMSO with water is an exothermic reaction, thus the buffer needs to be cooled on ice before being applied to the column (no DTT).
- HA buffer (5 liters): 5 mM KPO4, pH 7.6
- HB buffer (0.5 liter): 40mM KPO4 pH 7.6, 1.5M glycine, and 0.5 mM DTT
- Saturated ammonium sulfate (4 liters): Add about 1600 g of (NH4)2SO4 to 2 liters of water to make a saturated solution. Adjust to pH 7.6 with ammonia.
- Poly-L-proline (PLP) affinity matrix: For the preparation size described here, mix 0.5 g of poly-(L-proline) to 30 g of CNBr-activated Sepharose 4B for 2 h at room temperature or overnight at 4°C according to the supplier's recommendations. Avoid extensive stirring, as this will have deleterious effects on the matrix. Monitor the coupling reaction photometrically at 234 nm in supernatants of small samples of the incubation mixture, removed at intervals. After each use, wash the matrix immediately with 1 liter of water, followed by 1 liter of 1% N-lauryl sarcosine, followed with 1 liter of water. Reequilibrate with PLP buffer containing 0.01% sodium azide. Stored at 4°C, this matrix should be good for 10-15 preparations and last for 1-2 years.
- Hydroxyl apatite column: Degas 100ml of settled hypatite-C in 500ml buffer HA. Pack an XK-26/20 column with hydroxyl apatite at a flow rate of 10ml/min. Attach a top adaptor, but allow some buffer to be present on top of the matrix and equilibrate the hydroxyl apatite column with 1 liter of buffer HA at a flow rate of 10ml/min.
Steps
- Make 5 liters (1×) PLP buffer using the (2×) buffer stock. Degas all buffers.
- Partially thaw 700 g thymus and remove fat and connective tissue. Cut the organ into small pieces and homogenise in a blender using approximately 2 volumes of homogenisation buffer.
- Transfer the tissue extraxt to 500-ml polycarbonate bottles and centrifuge at 20,000g (12,000rpm with the JLA-16.250 rotor) for 30min.
- Filter the supernatant through glass wool, transfer to 70-ml polycarbonate bottles, and centrifuge at 100,000g (30,000rpm with the 45TI rotor) for 30 min.
- Prepare the PLP column in a laminar flow hood: Pack an XK-50/20 column with the affinity matrix described in step 6 of the previous section and equilibrate it with PLP buffer at a flow rate of 5-10ml/min.
- Filter the supernatant from step 4 through glass wool and load it onto the PLP column.
- After all the protein has been loaded, wash the matrix with PLP buffer until the color is removed from the matrix.
- Elute bound proteins with ice-cold PLP elution 0.4 buffer into a 500-ml measuring cylinder on ice. After this step the preparation already contains mainly profilin and profilin:actin complexes.
- Dilute the PLP eluate with 2 volumes of buffer HA and load the solution onto the hydroxyl apatite column at a rate of 10ml/min, monitoring the UV absorbance at 280nm. The DMSO accounts for a small increase in absorbance at 280nm. A tough layer may form on top of the matrix bed, restricting buffer flow. If necessary, stop the pump, disconnect the top adaptor, and resuspend the top centimeter of matrix bed by careful stirring with a glass pipette. Allow the matrix to resettle, replace the top adaptor, and continue loading.
- After the protein has entered the column, wash with buffer HA to remove DMSO. Finish washing when a low baseline is reestablished. Carefully press the top adaptor onto the gel matrix.
2. Separation of Profilin-Bound β- and γ-Actin Isoforms
- For separation of profilin: β-actin from profilin: γ-actin complexes, elute the hydroxyl apatite-bound material with a linear gradient of 5mM potassium phosphate to 40 mM phosphate/1.5 M glycine. Employ a gradient mixer containing 350ml HA in the start buffer compartment and 350ml HB in the finishing buffer compartment. (If a different column dimension is used, the gradient length should be approximately 12 column volumes.) Elute the protein at a flow rate of 0.3 ml/min. Collect 10-min fractions.
- Identify the fractions containing profilin: γ-actin and profilin: β-actin and pool the appropriate fractions (see Fig. 1). Continue as described in step 14.
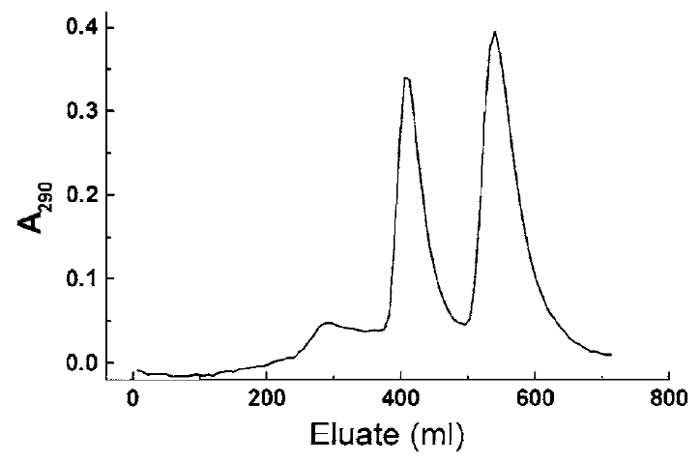 |
| FIGURE 1 Separation of profilin, profilin : β-actin, and profilin : γ-actin using hydroxyl apatite chromatography (see Section III,A). The slightly more basic γ-actin : profilin complex elutes at lower ionic strength, followed by the β-actin : profilin complex at higher phosphate/glycine concentration. The small initial peak contains excess profilin. |
- If isoform separation is not aspired, elute the hydroxyl apatite-adsorbed protein using HB buffer. The eluate consists of mixed actin isoforms in complex with profilin, plus a slight excess of profilin.
4. Ammonium Sulfate Precipitation of PA
- Precipitate the protein by dialysis against 2 liters of saturated ammonium sulfate using dialysis tubing with a molecular mass cutoff under 12 kDa. The resulting protein is suitably stored as a precipitate in ammonium sulfate. Stored in this form at 4°C the PA is stable for 6-12 months. Estimate the concentration of protein by diluting 25-50µl of the well-suspended solution into 1 ml of water and determine the absorbance at 280 and 310nm (280-310 roughly corresponds to milligram per milliliter).
The dialysis against saturated ammonium sulfate results in a reduction of the sample volume, ensuring effective protein precipitation. It is important to use enough saturated ammonium sulfate to avoid dilution of the salt below 40-50% saturation, which is the concentration precipitating the profilin: actin.
For efficient extraction of the tissue, it may be helpful to pass the organ through a meat grinder before homogenisation in a blendor. The buffer over tissue ratio should not be lower than 2:1. Continue homogenisation until no chunks are visible, which should take about 2 to 3min. Do not continue homogenisation over prolonged periods of time. Avoid excessive foaming.
5. Isolation of Profilin and Actin from Precipitated P :A Complex
Solutions and Columns
- G buffer (2 liters): 5 mM Tris-HCl, pH 7.6, 0.5 mM ATP, 0.1 mM CaCl2, and 0.5 mM DTT
- Resuspension buffer (0.1 liter): G buffer, add DTT to 5mM
- F buffer (0.1 liter): 5 mM Tris-HCl, pH 7.6, 0.5 mM ATP, 2 mM MgCl2, 50 mM KCl, and 0.5 mM DTT
- 2 M KP04 pH 7.6 (make 50ml)
- P buffer (1 liter): 10 mM KPO4 pH 7.6, and 0.5 mM DTT
- Prepare an XK-26/70 Sephacryl-300 gel filtration column equilibrated with G buffer
- Prepare an XK-26/50 Sephadex G-25 gel filtration column equilibrated with P buffer
- Prepare an XK-16/10 DEAE-Sephadex anionexchange column equilibrated with P buffer
6. Purification of Actin
- Collect 30-50mg of ammonium sulfateprecipitated profilin:actin (either isoform; previous section, step 14) by centrifugation in a JLA-16.250 rotor at 10,000rpm for 10 min. Discard the supernatant, leave the tubes upside down for a few minutes, and then wipe the inside of the tubes with Kleenex to remove residual ammonium sulfate.
- Dissolve pellets in a small volume of resuspension buffer, stir, and add buffer until the solution becomes clear. Protein concentration should be 10- 20mg/ml. Centrifuge the solution again (as in step 1) to remove undissolved residues. Save the supernatant.
- Measure the volume of the protein solution and add MgCl2 to 5 mM and EGTA to 0.5 mM. Allow actin to polymerise at room temperature for 30min.
- Induce paracrystal formation by the addition of 1 volume of 2M KPO4. Mix carefully; avoid extensive pipetting. Leave for 1 h at room temperature or overnight at 4°C.
- Collect the actin paracrystals by centrifugation at 10,000g for 20min at 15°C. The supernatant contains profilin and small amounts of remaining profilin: actin; save supernatant on ice for the profilin purification described later. The pellet contains actin paracrystals.
- Resuspend the paracrystals in F buffer to dissolve the actin filaments. Mix by careful pipetting until all lumps have dissolved.
- Sediment filaments by ultracentrifugation at 100,000g for 3 h at 15°C. Remove the supernatant and add a small amount of G buffer to the F-actin pellet and leave it for 10-20min. Seal the tip of a Pasteur pipette and use it to scrape the pellet off the walls of the tube. Carefully transfer the pieces of F-actin gel to a Dounce-type glass homogeniser and homogenise without introducing air. Measure the protein concentration using an extinction coefficient at A290-310 of 0.63 ml/mg × cm. Dilute the F-actin to 5 mg/ml to get efficient depolymerisation.
- Dialyze the actin sample against 10 volumes of G buffer in 12- to 14-kDa cutoff tubing with three buffer changes and homogenisations. Four to 6 h of dialysis is sufficient for every buffer change.
- Subject the dialyzed actin to ultracentrifugation at 100,000g for 3 h at 4°C in order to remove undissolved material. Load the supernatant onto the Sephacryl S-300 column. The sample volume should not exceed 4% of the column volume. Elute with G buffer at a flow rate of 30ml/h. Collect 4- to 6-ml fractions. Measure protein concentration in the fractions and pool the protein peak. Analyze by SDS-PAGE, drop freeze (20 µl droplets) the actin in liquid N2, and store in a nitrogen tank.
7. Purification of Profilin
- Desalt the profilin solution from step 5 by gel filtration over Sephadex G-25. The volume of the protein solution may be up to approximately 30% of the column bed volume. Collect 2- to 3-ml fractions.
- Determine protein concentrations across the chromatogram. Measure conductivity in the last fractions to make sure the solution will not be contaminated by salts and pool the protein-containing fractions. 12. Run the G-25 pool over the small DEAESephadex column. Contaminating actin will stay on the ion exchanger. The column flow through contains pure profilin. Quick-freeze aliquots in liquid N2 and store at -80°C.
Before inducing paracrystal formation (step 4), check on the progress of the polymerisation reaction by rocking the tube carefully; the protein solution should have a gel-like consistence. If necessary, stimulate polymer formation by a few short bursts in an ultrasonicator.
Polymers of cytoplasmic actin are more cold sensitive than polymers of α-actin. Therefore, the ultracentrifugation step that aims at collecting actin filaments is carried out at 15°C.
B. Purification of Recombinant β-Actin from Baker's Yeast
This protocol is used for the large-scale preparation of recombinant actin expressed in Saccharomyces cerevisiae strain K923 using the temperature-inducible expression system described by Karlsson (1988). The heterologous actin cDNA is placed under the combined control of the PGK promoter and the negative transcription factor α2 and is introduced into strain K923 containing a temperature-sensitive mutation in the mating type switch (Walton and Yarranton, 1989; see also Sledziewski et al., 1988). This results in temperature- inducible expression of the heterologous actin. At 34°C, the cells are MATα and produce the α2 protein, which blocks transcription of the cloned cDNA. By lowering the temperature to 23°C, the cells switch mating type and cease to produce α2, leading to expression of the recombinant actin. Total actin is isolated from yeast extracts by DNase I affinity chromatography (Zechel, 1980), and the recombinant isoform is separated from the endogenous actin by hydroxyl apatite chromatography (Karlsson, 1988). Ten liters of flask culture produces 120-150g of yeast cells (the amount needed for a preparation as described later), yielding in the range of 10-15mg each of yeast and recombinant actin. Usually, a fermentor culture in 10-liter fermentors is used, producing 700-800 g of yeast, which is material for five to six standard preparations of recombinant actin. The fermentation process has successfully been scaled to 50-100 liters. It should be noted that the yeast does not modify the N terminus of actin and does not methylate residue His73 (for discussion, see Nyman et al., 2001).
This method has been adapted for the production of human profilin (Aspenström et al., 1991) and should also work for other proteins.
Solutions
- S. cerevisiae strain K923: HMLα, mat::LEU2+ hmr ::TRP1+ ura3, ade2, sir3ts; MATa at 23°C, MATα at 34°C. This strain is grown on standard YPD medium (Sambrook and Russell, 2001) at 34°C.
- UYM media for transformed yeast strain K923: Bottle 1:8 g yeast nitrogen base without amino acids, 55 mg adenine sulfate, 55 mg L-Tyr; add H2O to 580 ml. Bottle 2:11 g casamino acid vitamin assay; add H2O to 300ml. Autoclave, allow to cool. Combine contents of bottles 1 and 2 and add 100ml 20% glucose, 10ml 0.5% L-Trp, and 10ml 1% L-Leu. Grow the transformed strain on min-ura plates at 34°C.
Steps
- Culture K923 transformed with pY-β-actin to single colonies on a min-ura plate at 34°C. Inoculate into 10ml UYM and culture for 24h under vigorous shaking at 34°C, transfer into 1 liter of UYM, and continue culturing for another 24 h at 34°C.
- Transfer the inoculum into the fermenter, which has been prepared with 9 liters modified YPD [220g peptone, 220 g yeast extract, 2.2 g adenine sulfate, 2.5 g glucose (0.25%), and PPG to avoid foaming]. Glucose is added continuously from a 50% stock, adjusting the concentration with the density of the culture (measured by OD640) to keep it at approximately 0.5%. Approximately 12h after inocculation, add 220g of yeast extract in 500ml water. Keep the culture at 34°C until OD640 is between 1 and 2 (this takes 6-8 h). At this stage, lower the temperature to 23°C to initiate the mating-type switch, which induces production of the recombinant protein. If larger scale (50-100 liters) fermenations are required, prepare the inoculum in the fermentor at a volume of 5-10 liters in UYM medium before transferring into a bigger fermentor with medium as described earlier.
- Harvest the culture when OD640 no longer increases (ca. 24-26 h after inocculation). Rapidly cool the culture to below 10°C, start collecting it into ice-cold l-liter centrifuge flasks, and centrifuge at 5000rpm for 20 min (Sorvall RC-3 or equivalent). Collect all material into one or two flasks, centrifuge at 5000 rpm for 30 min, aliquot into suitable portions (150 g) for preparation of the protein, and store at -70°C.
Solutions and Columns
- 2× G buffer (3 liters): 10mM Tris-HCl, pH 7.6, 0.2 mM CaCl2, and 1 mM ATP
- G buffer: use 2.5 liters of the 2xG buffer to make 5 liters (5 mM Tris-HCl, pH 7.6, at 4°C, 0.1 mM CaCl2, and 0.5 mM ATP)
- Na-acetate buffer (0.5 liter): 0.5M sodium acetate, 0.5 mM ATP, pH 7.6, 0.1 mM CaCl2, 10% glycerol, and 0.5 mM DTT
- DNase wash buffer Elu-I: use the 2×G buffer to make 0.5 liter Elu-I: 5 mM Tris-HCl, pH 7.6, at 4°C, 0.1 mM CaCl2, 0.5 mM ATP, 10% formamide, 10% glycerol, and 0.5 mM DTT
- DNase elution buffer Elu-II: Use the 2×G buffer to make 0.3 liter Elu-II: 5 mM Tris-HCl, pH 7.6, at 4°C, 0.1 mM CaCl2, 0.5 mM ATP, 40% formamide, 10% glycerol, and 0.5 mM DTT
- HA buffer (0.5 liter): 5mM KPO4, pH 7.6, and 0.5 mM DTT
- HB buffer (100 ml): 40 mM KPO4, pH 7.6, 1.5M glycine, and 0.5 mM DTT
- Water-diluted protease inhibitor mix (1000 × stock): 0.5mg/ml each of antipain, leupeptin, pepstatin A, chymostatin, and aprotinin. Store 0.5-ml aliquots at -20°C.
- Ethanol-diluted protease inhibitor mix (100 × stock): 0.1M phenylmethyl-sulfonyl fluoride (PMSF) 1 mM benzamidine-HCl, and 0.1mg/ml phenanthroline. Store 5-ml aliquots at -20°C.
- RNase A (500 × stock): 10mg/ml in 0.1M sodium acetate, pH 5.0. Boil in a water bath for 20min. Store 1-ml aliquots at -20°C.
- ATP for Hypa fractions: 10mM ATP in 40mM K2HPO4; gives a pH of approximately 7.0
- D buffer (1 liter): 10mM Tris-HCl, pH 7.6, 150mM NaCl, 0.5 mM CaCl2, and 0.01% sodium azide
- DNase I affinity matrix: Couple 0.3 g of DNase I to 15g of CNBr-activated Sepharose 4B overnight at 4°C according to the supplier's recommendations. Include 1 mM CaCl2 in all buffers. Monitor the coupling reaction photometrically at 280 nm (see step 6 of Section III,A). According to our experience, this matrix will have a capacity of approximately 40mg actin and will allow purification of actin from up to 150g (wet weight) of yeast coexpressing recombinant actin. Store the matrix in D buffer containing 0.01% sodium azide. If column capacity decreases after several preparations, wash off bound contaminants with 4M guanidine hydrochloride, 0.5M sodium acetate, and 30% glycerol. Kept at 4°C, this matrix should be good for 10-15 preparations and last for 1-2 years. Also note that long-term exposure of DNase I to reducing agents inactivates the enzyme.
- Hydroxyl apatite column: Degas 20ml of settled hypatite C. Pack an XK-16/20 column with the matrix and equilibrate with HA buffer at 120ml/h.
Steps
- Use up to 150g (wet weight) of yeast cells (corresponding to 10-12 liters of flask culture). Make 500ml lysis buffer by adding water-diluted and ethanol-diluted protease inhibitors and 50 gl polypropylene glycol. Thaw the pelleted yeast in 100ml lysis buffer. Lyse the yeast cells by passage through the bead mill and wash out the lysate using the remaining lysis buffer.
- Add RNase A. Centrifuge the collected yeast lysate in a large-volume rotor at 12,000rpm for 40min at 4°C. Carefully decant the supernatant and filter it through glass wool.
- Pour an XK-50/20 column using the DNase affinity matrix (step 13 of the previous section). Equilibrate the DNase column with G buffer. Pump the yeast lysate onto the DNase column. Wash with G buffer until a stable baseline is established as judged by OD290. Wash consecutively with 5 column volumes of Na-acetate buffer, wash buffer Elu-I, and G buffer.
- Prepare 200ml of dilution buffer (G buffer containing 10% glycerol). Arrange a measuring cylinder with 100 ml dilution buffer on a magnetic stirrer. Blank a spectrophotometer with buffer Elu-II. Reduce the pump flow rate and start elution with buffer Elu-II. Follow the A290; at 0.05-0.1, start collecting into the dilution buffer. The volume of dilution buffer should be at least 50%. Add more dilution buffer if necessary. Stop collecting when the A290 of the eluate is below 0.1.
- Apply the diluted actin eluate onto the hydroxyl apatite column at a flow rate of max 100ml/h, i.e., somewhat slower than the packing flow rate, to avoid further packing of the matrix with the more viscous actin eluate.
- Prepare a fraction collector with 3-ml tubes containing 100-gl aliquots of buffered 10mM ATP (for a final ATP concentration of 0.5 mM). Start collecting 2- ml fractions. Elute the actin isoforms using a gradient of 60ml each of buffers HA and HB at a flow rate of 10ml/h.
- Identify actin-containing fractions by measuring the absorbance at 290nm (Fig. 2), DNase I inhibition activity, SDS-PAGE, or dot blotting. Pool the fractions and dialyse against G buffer; alternatively, the pools may be concentrated and subjected to gel filtration.
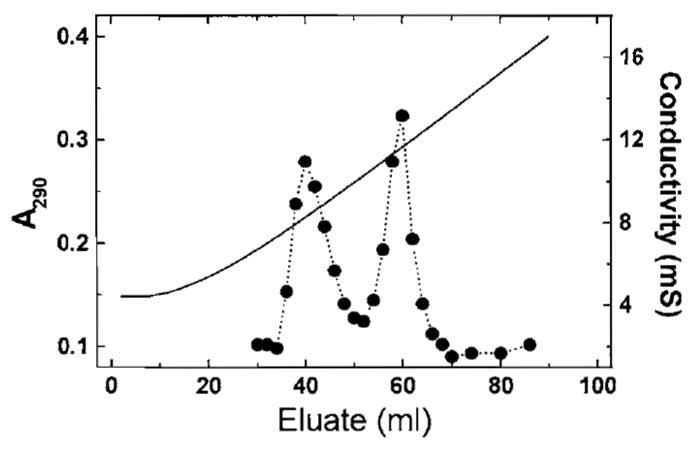 |
| FIGURE 2 Separation of yeast actin and recombinant nonmuscle β-actin using hydroxyl apatite chromatography (Section III,B). The more acidic β-actin elutes at the higher phosphate/glycine concentration. Solid line represents the conductivity. |
The protease inhibitor PMSF is counteracted by reducing agents. Therefore, DTT should be excluded from the lysis buffer and added to buffers only after the yeast extract has passed over the DNase column.
Failure to wash the DNase affinity column with high ionic strength (sodium acetate) buffer will result in contamination by a number of actin-binding proteins, with the most prominent one being yeast cofilin.
Prolonged exposure to 20% formamide will lead to actin denaturation. Therefore, it is important to collect the actin eluate into at least an equal volume of dilution buffer and apply it to the next column as quickly as possible. Attempts to store quick-frozen actin in this buffer have been unsuccessful.
The most probable reason for poor isoform separation is too high a flow rate during elution.
The aforementioned conditions for the separation of recombinant β-actin from yeast actin are not necessarily applicable to mutants of β-actin or other recombinant actins (e.g., Aspenstr6m and Karlsson, 1991). Therefore, it may be necessary to alter conditions.
References
Aspenström, P., and Karlsson, R. (1991). Interference with myosin subfragment-1 binding by site-directed mutagenesis of actin. Eur. J. Biochem. 200, 35-41.
Aspenstr6m, P., Lassing, I., and Karlsson, R. (1991). Production, isolation and characterization of human profilin from Saccharomyces cerevisiae. J. Muscle Res. Cell Motil. 12(2), 201-207.
Gorbunoff, M. J. (1990). Protein chromatography on hydroxyapatite columns. Methods Enzymol. 182, 329-339.
Innocenti, M., Zucconi, A., Disanza, A., Frittoli, E., Areces, L. B., Steffen, A., Stradal, T. E., Di Fiore, P. P., Carlier, M. R, and Scita, G. (2004). Abil is essential for the formation and activation of a WAVE2 signalling complex. Nature Cell Biol. 6, 319-327.
Jazwinski, S. M. (1990). Preparation of extracts from yeast. Methods Enzymol. 182, 154-174.
Karlsson, R. (1988). Expression of chicken beta-actin in Saccharomyces cerevisiae. Gene 68, 249-257.
Lindberg, U., Schutt, C. E., Hellsten, E., Tj/ider, A. C., and Hult, T. (1988). The use of poly(L-proline)-Sepharose in the isolation of profilin and profilactin complexes. BBA 967, 391-400.
Nyman, T., Schiller, H., Korenbaum, E., Schutt, C. E., Karlsson, R. and Lindberg, U. (2002). The role of MeH73 in actin polymerization and ATP hydrolysis. J. Mol. Biol. 317(4), 577-589.
Pardee, J. D., and Spudich, J. A. (1982). Purification of muscle actin. Methods Enzymol 85, 164-181.
Ridley, A. J., Schwartz, M. A., Burridge, K., Firtel, R. A., Ginsberg, M. H., Borisy, G., Parsons, J. T., and Horwitz, A. R. (2003). Cell migration: Integrating signals from front to back. Science 302, 1704-1709.
Segura, M., and Lindberg, U. (1984). Separation of non-muscle isoactins in the free form or as profilactin complexes. JBC 259(6), 3949-3954.
Selden, L. A., Kinosian, H. J., Estes, J. E., and Gershman, L. C. (2000). Cross-linked dimers with nucleating activity in actin prepared from muscle acetone powder. Biochemistry 39, 64-74.
Sledziewski, A. Z., Bell, A., Kelsay, K., and MacKay, V. L. (1988). Construction of temperature-regulated yeast promoters using the MATα2 repression system. Bio/Technology 6, 411-416.
Walton, E. E, and Yarranton, G. T. (1989). Negative regulation of gene expression by mating-type. In "The Molecular and Cell Biology of Yeasts" (E. E Walton and G. T. Yarranton eds.), pp. 43-69. Blackie, Glasgow, UK.
Zechel, K. (1980). Isolation of polymerization-competent cytoplasmic actin by affinity chromatography on immobilized DNase I using formamide as eluant. Eur. J. Biochem. 110, 343-348
Support our developers




