Purification of Smooth Muscle Actin
Actin purified from mammalian or avian skeletal muscle is a readily available probe to study the potential interaction of a putative actin-binding protein or recombinant fragment in vitro. Yet, the use of a specific actin isotype for the determination of binding affinities or the tissue-specific regulatory potential of actinassociated proteins is often required.
Purification of actin from smooth muscle is much less straightforward than from skeletal muscle tissue. The major difficulty resides in the large number of actin-binding proteins, which populate the actin filaments in this tissue type. These proteins interfere with polymerization even at levels undetectable on conventional Coomassie blue-stained SDS-PAGE gels. A primary source of contamination is the copurification of tropomyosin. Moreover, because the total amount of actin present in smooth muscle is reduced compared to that in skeletal muscle, the yield is lower. However, despite the aforementioned potential pitfalls, smooth muscle actin can be purified in large quantities and high purity by the use of larger extraction volumes in the initial steps.
This article describes the method used for purifying polymerization-competent actin from mammalian or avian visceral smooth muscle (porcine stomach or turkey gizzard) and suggests some useful methods to test the functionality of the preparation prior to its use in analytical assays. The purification scheme follows the original modification of Sobieszek and Bremel (1975). The modified extraction buffer (containing 10 mM imidazole, 10 mM bis-Tris, 40 mM KCl, 2 mM MgCl2, 1 mM cysteine, and 5mM EGTA at pH 6.5) introduced by Fiirst (1986) significantly improved the solubilization and removal of actin-binding/modulating proteins.
Chemicals ATP (Sigma, Mr 551.10, Cat. No. A-3377); bis-Tris (Fluka, Mr 209.24, Cat. No. 14880); cysteine (Sigma, Mr 175.60, Cat. No. C-7880); CaCl2 (Merck, Mr 147.02, Cat. No. 2382); DTE (Serva, Mr 154.30, Cat. No. 20697); EGTA (Sigma, Mr 380.40, Cat. No. E-3889); imidazole (Merck, Mr 68.08, Cat. No. 1.04716); KCl (Merck, Mr 74.56, Cat. No. 1.04936); MgCl2 (Merck, Mr 203.30, Cat. No. 5833); NaCl (Merck, Mr 58.40, Cat. No. 1.06404); sucrose (Fluka, Mr 342.39, Cat. No. 84-100); Tris (ICN, Mr 121.14, Cat. No. 819 623); and ammonium sulphate (Fluka, Mr 132.14, Cat. No. 09982)
Gel Filtration Media
Sephadex G-150 or Sephacryl S100HR (Amersham Biosciences)
III. SOLUTIONS
- G-actin buffer: 2 mM Tris-HCl, pH 8.0, 0.1 mM CaCl2, 0.2 mM ATP, and 0.1 mM DTE
- F-actin buffer: 2 mM Tris-HCl, pH 7.4, 150mM KCl, 2 mM MgCl2, and 2 mM EGTA
- 10x polymerization stock: 1.5M KCl, 20mM MgCl2, and 20 mM EGTA
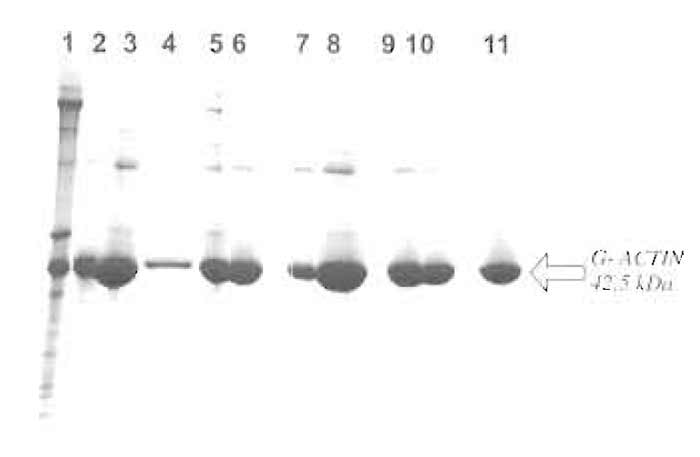 |
| FIGURE 1 Commassie blue-stained 8-23% gradient SDS-PAGE gel showing the individual steps of the purification procedure. Lane 1, acetone powder extract; lane 2, supernatant after centrifugation; lane 3, 25% ammonium sulphate pellet; lane 4, supernatant after 25% ammonium sulphate; lane 5, precipitate after dialysis against G-actin buffer; lane 6, G-actin after dialysis and centrifugation; lane 7, supernatant after polymerization; lane 8, polymerized F-actin; lane 9, pellet after 48 h of dialysis against G-actin buffer; lane 10, G-actin after dialysis, applied to column; and lane 11, peak fraction from Sephacryl S-100 HR column (pure smooth muscle actin). |
The most convenient source of actin for relatively large yields is the powder remaining after the extensive drying of muscle homogenates in acetone. For this procedure, prepare the acetone powder from chicken gizzard smooth muscle according to Strzelecka- Golaszewska et al. (1980).
Starting with 15 g of dry acetone powder, the extraction in a final volume of 1.3 liters of G-actin buffer is let to proceed for 20-25 min at 4°C under constant agitation (using a magnetic stir bar). Centrifuge the resulting extract at 15,000g for 60min (Sorvall GS-A rotor or equivalent).
From the supernatant, precipitate actin by the slow addition of solid ammonium sulphate to reach 25% saturation. Let the precipitation proceed for 90 minutes at 4°C. Collect the precipitate by centrifugation at 15,000g for 30min (Sorvall GS-A rotor or equivalent).
Resuspend the pellet in about 50ml G-actin buffer and dialyze against 2 liters of G-actin buffer overnight with two to three changes of buffer. Centrifuge the dialyzed actin solution to remove polymerized or aggregated actin at 32,000g for 60min (Sorvall SS-34 rotor or equivalent).
From this clarified solution, polymerize F-actin by the addition of 150mM KCl, 2mM MgCl2, and 2mM EGTA (final concentrations) by adding 1 / 10 volume of 10x polymerization solution. Allow polymerization to proceed for 90min at 25°C.
Dialyze against 2 liters of G-actin buffer for 48h with four to six changes of dialysis solution to thoroughly depolymerize the F-actin. Remove precipitated or polymerized material by centrifugation at 32,000g as described earlier.
Measure the final concentration of the G-actin solution using the absorption coefficient A1%/290 of 6.5. Aliquots can be stored at -70°C after adding 2mg sucrose/mg of G-actin from a 600-mg/ml stock solution.
V. TESTS
There are several possibilities to test the functionality of the actin solution before using it in in vitro assays.
A. General Polymerization
Polymerize the G-actin solution as described earlier (by adding 1/10 volume of the 10× polymerization stock solution) and centrifuge the polymerized actin at 100,000g for 30min. Analyze the supernatant and pellet on a Coomassie blue-stained SDS-PAGE gel. The supernatant should essentially be devoid of actin, indicating 100% polymerization competence of the G-actin solution.
B. Cosedimentation Assays
Polymerize the G-actin solution as described earlier, but in the presence of actin-binding proteins such as α-actinin and/or tropomyosin, and analyze supernatants and pellets by SDS-PAGE. The presence of the actin-binding protein in the pellet indicates a "functional surface" and intact binding sites on the actin filament.
VI. COMMENTS
We found that dry acetone powder can be stored for at least 20 years at -20°C without a significant loss of quality of the purified actin. It thus may pay to stock up on acetone powder.
Initial extraction beyond the recommended 25 min results in the increased extraction of tropomyosin, which is difficult to remove at later stages. Accepting a lower yield is rewarded with higher purity!
The choice of buffer for the assays appears critical. F-actin reacts with structural alterations to different ionic strengths and the presence or absence of divalent or monovalent cations. The illuminating work by the Egelman group is a rewarding read.
References
Belmont, L. D., Orlova, A., Drubin, D. G., and Egelman, E. H. (1999). A change in actin conformation associated with filament instability after Pi release. Proc. Natl. Acad. Sci. USA. 96, 29-34.
Fürst, D. O. (1986). Ph.D. Thesis, University of Salzburg.
Galkin, V. E., Orlova, A., Lukoyanova, N., Wriggers, W., and Egelman, E. H. (2001). Actin depolymerizing factor stabilizes an existing state of F-actin and can change the tilt of F-actin subunits. J. Cell Biol. 153, 75-86.
Orlova, A., and Egelman, E. H. (1993). A conformational change in the actin subunit can change the flexibility of the actin filament. J. Mol. Biol. 232, 334-341.
Sobieszek, A., and Bremel, R. D. (1975). Preparation and properties of vertebrate smooth-muscle myofibrils and actomyosin. Eur. J. Biochem. 55, 49-60.
Strzelecka-Golaszeska, H., Prochniewicz, E., Nowakl, E., Zmorsynski, S., and Drabikowski, W. (1980). Chicken gizzard actin: Polymerization and stability. Eur. J. Biochem. 104, 41.
Support our developers




