Genome-Wide Screening of Intracellular Protein Localization in Fission Yeast
I. INTRODUCTIONIntracellular localization is an important part of the characterization of a gene product. In order to search for genes based on the intracellular localization of their products, we constructed a green fluorescent protein (GFP)-fusion genomic DNA library of the fission yeast Schizosaccharomyces pombe (Ding et al., 2000). This was accomplished by fusing random fragments of genomic DNA to the 5' end of the GFP gene in such a way that the expression of potential GFP-fusion proteins would be under the control of the endogenous promoters contained in the genomic DNA fragments. The intracellular localization of the fusion proteins was determined by microscopic observation of individual transformants. As genes fused to GFP are expressed under their native promoters, the intracellular localization of gene products can be examined under physiological conditions in both mitotic and meiotic cells. This library provided the foundation for a survey of the intracellular localization of S. pombe proteins. Now that the S. pombe genome project is completed, genes can be searched from the intracellular locations of their protein products using our image database. Our library of GFP-fusion constructs also provides useful fluorescent markers, which can be used readily for microscopic observation in living cells and for various intracellular structures and cellular activities.
II. MATERIALS AND INSTRUMENTATION
Polypeptone is from Nihon Pharmaceutical (Cat. No. 394-00115), yeast extract is from Difco (Cat. No. 212750), agar is from Wako (Cat. No. 010-15815), ampicillin is from Wako (Cat. No. 016-10373/TWK6416), glucose is from Wako (Cat. No. 041-00595), Zymolyase- 100T is from Seikagaku (Cat. No. 120493/109406), novozym 234 is from Novo Nordisk (Cat. No. 2880/PPM4356), proteinase K is from Merck (Cat. No. 24658-0100/V354968 92), and T4 DNA ligase is from TaKaRa (Cat. No. 2011A/1511). Water used for the preparation of the culture medium and all of the solutions is first treated using the Elix 5 water purification system (Millipore) and then the MilliQ ultrapure water purification system (Millipore).
Ten-well 25 x 75-mm glass slides are from Polyscience (Cat. No. 18357), and glass-bottom culture dishes are from MatTek (Cat. No. P35G-1.5-10-C). The DNA sequencer used is the ABI377 from Perkin-Elmer. A computer-controlled, fluorescence microscope system (DeltaVision, Applied Precision; Haraguchi et al., 1999) is used for visual screening of the GFP library. This microscope system is based on an inverted fluorescence microscope (IX70, Olympus Optical) equipped with a Peltier-cooled, charge-coupled device (CCD) (CH250 or CH350, Photometrics Ltd., Tucson, AZ). The objective lens used is an Olympus oil immersion lens (SPlan Apo 60/NA 1.4).
III. PROCEDURES
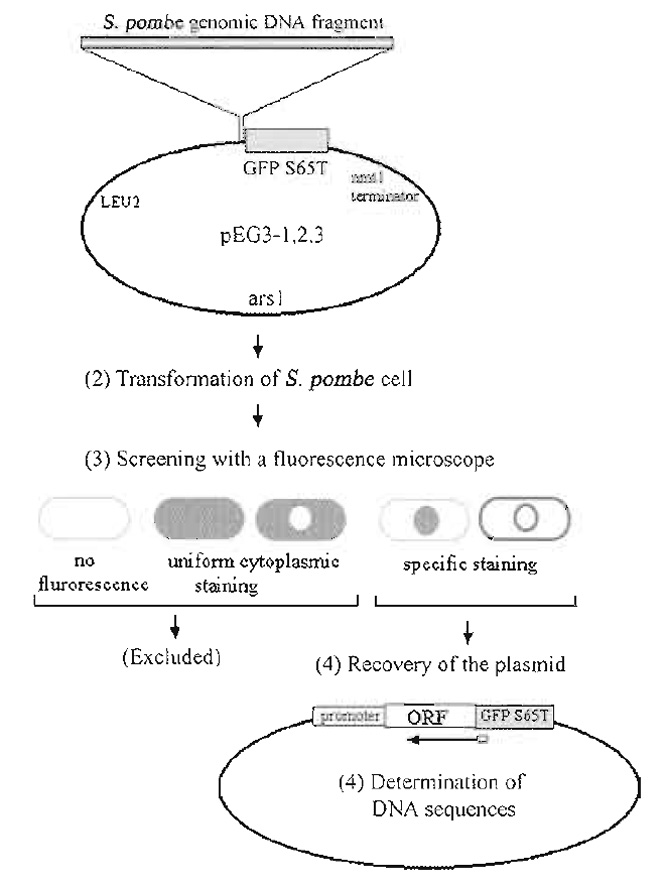 |
| FIGURE 1 Strategy for construction and screening of the GFP-fusion genomic DNA library. Modified from Ding et al. (2000) |
A. Construction of the GFP-Fusion Genomic DNA Library
Figure 1 summarizes our strategy for the construction and screening of a gene library in which S. pombe genomic DNA fragments from a wild-type strain were fused to the 5' end of the GFP-S65T gene.
1. Construction of Plasmid Vectors for the Library
The construction of one such library is described in detail in Ding et al. (2000). The multicopy plasmid used as a vector for our library was created by modification of a popular vector for fission yeast, pREP1 (Maundrell, 1993). In order to express GFP-fusion genes under the control of endogenous yeast promoters, the nmtl promoter region in pREP1 was deleted; therefore, only genes in DNA fragments that also contained the genes' endogenous promoters would be expressed. Next, the coding sequence of GFP-S65T (Heim and Tsien, 1996) was amplified by polymerase chain reaction (PCR), and inserted upstream of the nmtl terminator. Linker oligonucleotides were then designed such that all three reading frames of the restriction DNA fragments were fused to the GFPcoding sequence. Each of the resultant plasmids contained the S. pombe arsl as a replication origin and the S. cerevisiae LEU2 gene as a selection marker, both of which were derived from the pREP1 plasmid (Fig. 1).
Insertion of the GFP PCR product into the nmtl promoter deleted pREP1, resulting in pEG3-1. This plasmid has a BamHI site immediately 5' to GFP and was used as the first frame library plasmid. The second frame plasmid, pEG3-2, was constructed by inserting a 12 base (CCCAGATCTGGG) BglII linker into pEG3-1 that had been digested with BamHI and blunt-ended. The third frame plasmid, pEG3-3, was constructed by inserting a 10 base (CCAGATCTGG) BglII linker into BamHI digested, blunt-ended pEG3-1. Importantly, restriction enzyme sites that can be used for recloning of the yeast gene into other GFP-fusion vectors must be present. In our library, DNA restriction fragments were cloned into the BamHI site of pEG3-1. In pEG3-2 and pEG3-3, however, the BamHI site was absent, as the BglII linkers were blunt ended into this site. Therefore, the BamHI site present in pEG3-1 was used to reclone genes from this plasmid, while BglII restriction sites were used to reclone genes present in pEG3-2 and pEG3-3.
Insertion of the library of DNA restriction fragments into each of the three plasmids, pEG3-1, pEG3-2, and pEG3-3, resulted in a library of random fragments of genomic DNA fused to the GFP-coding sequence. Microscopic observation was used to eliminate transformants that did not express GFP-fusion proteins (Fig. 1). These included transformants that did not express a GFP-fusion protein due to the absence of an endogenous promoter to drive the expression of the fusion gene or because the GFP-coding sequence was not in-frame with the fission yeast protein-coding sequence. Thus, after visual screening, a significant proportion of the library consisted of fission yeast genes fused in-frame with GFP and driven by their natural biological promoters.
An important point that needs to be taken into consideration in this type of library is that the linker oligonucleotides used to generate alternative GFP reading frames introduce a spacer between GFP and the yeast gene, and this spacer may affect the localization of the GFP-fusion protein (see Section IV).
2. Preparation of Genomic DNA
The S. pombe strain 968 h90 was used to prepare genomic DNA. Genomic DNA was isolated according to Matsumoto et al. (1987), which involves two centrifugation steps for the isolation of nuclei to reduce mitochondrial DNA contamination.
Solutions
- YEade culture medium: 0.5% yeast extract, 3% glucose, 75µg/ml adenine sulfate dihydrate
- CPS buffer: 50mM citrate/phosphate, pH 5.6, 40mM EDTA, 1M sorbitol
- 10mM TE buffer: 10mM Tris-HCl, pH 7.5, 1 mM EDTA
- 50mM TE buffer: 50mM Tris-HCl, pH 7.5, 50mM EDTA
Steps
- Prepare a 200-ml liquid culture of S. pombe cells in a rich medium (YEade) and grow to midlog phase (1 × 107 cells/ml) with shaking at 33°C.
- Harvest by centrifugation at 2500rpm for 5 min.
- Wash cells in 10 ml of 10 mM TE buffer. Centrifuge at 2500rpm for 5 min.
- Resuspend cells in CPS buffer at 3.4 x 108 cells/ml, add Zymolase-100T and Novozym to a final concentration of 0.2mg/ml, and incubate at 36°C with gentle shaking for 30min to digest the yeast cell walls.
- Check digestion of cell walls using a phasecontrast microscope. Continue the digestion until 80% of the cells become round-shaped protoplasts.
- Spin down at 2000rpm for 5 min.
- Wash with 5 ml CPS buffer and resuspend in 5 ml ice-cold 10 mM TE buffer containing 1% Triton X-100.
- Spin down at 2000rpm for 5 min at 0°C. Transfer the supernatant to a new tube and centrifuge at 10,000rpm for 30min at 4°C. Discard the supernatant and dry the pellet.
- Resuspend the pellet in 5 ml of 50mM TE buffer. Add 0.5 ml 10% SDS, 0.375 ml 4M sodium chloride, and 0.5 mg protease K and incubate overnight at 50°C.
- Add 5ml of phenol/chloroform (1:1) and mix well. Spin down at 10,000 rpm for 10 min.
- Transfer the upper aqueous phase to a new tube and repeat step 10.
- Transfer the upper aqueous phase to a new tube and precipitate DNA with 2 volumes of ethanol. Wash the pellet with ice-cold 70% ethanol and dry under vacuum.
- Finally, resuspend DNA in 0.1ml of 10mM TE buffer containing 100µg/ml RNase A. The DNA can be quantified by running an aliquot on an agarose gel alongside a calibrated DNA sample. Typical yields are about 5-15µg per a starting culture of 200ml at midlog phase.
3. Partial Digestion of Genomic DNA
Genomic DNA is partially digested with a four-base cutter restriction enzyme, e.g., Sau3AI (Ding et al., 2000). A combination of several four- or six-base cutter restriction enzymes is also applicable (Sawin and Nurse, 1996). Considering the average size of fission yeast genes, including their promoters, 3- to 6-kb DNA fragments are appropriate. A suitable digestion time should be determined empirically in a small-scale pilot experiment. Centrifuge the partially digested DNA through a sucrose gradient (10-40%) at 50,000rpm for 15min using a RP-50T2 rotor in a Hitachi ultracentrifuge (Himac CP70G). Collect fractions and run aliquots out on an agarose gel. Pool the fractions that contain DNA fragments with the preferred length. Recover the DNA by ethanol precipitation.
4. Construction of Plasmid Libraries in Escherichia Coli
Digest each plasmid vector-when constructing our library, these were pEG3-1, pEG3-2, and pEG3-3mwith an appropriate restriction enzyme and treat with alkaline phosphatase to inhibit self-ligation of the plasmid. Run small-scale pilot ligation experiments to find ligation conditions that minimize background transformants derived from self-ligation. In our large-scale experiments, 30 ng of the alkaline phosphatase-treated vector and 150ng of DNA fragments were incubated with 350 units of T4 ligase in a total volume of 30µl overnight at 16°C. Competent E. coli (DH5α) were transformed with 30µl of each of the three plasmid DNA libraries and were plated on LB plates containing 50µg/ml ampicillin at a dilution suitable for the appearance of isolated single colonies. This procedure yielded approximately 12,000 E. coli transformants for each library. Ampicillin-resistant colonies were scraped from plates with LB medium. A portion of this pool of cells (1/10 volume) was saved in small aliquots at -80°C as a frozen stock. DNA was prepared from the remaining cells. Small-scale preparations of DNA from 30 individual transformants showed that 76-85% of the plasmids had insert DNAs with an average length of 5 kbp.
B. Transformation of S. pombe Cells with the Library
Three sets of transformation were carried out using the libraries based on the three plasmid vectors, pEG3- 1, pEG3-2, and pEG3-3. Homothalic S. pombe strains CRL126 (h90 leul-32 ura4 his2) and CRL152 (h90 leul- 32 ura4 lysl) were used for transformation with the GFP-fusion genomic DNA libraries. Homothalic (h90 strains can readily be induced to undergo meiosis and thus can be conveniently screened for mitotic and meiotic localization of fluorescently labeled proteins. A standard lithium chloride method (Moreno et al., 1991) can be used for the transformation of fission yeast cells with the library DNA. Transformants were selected on plates of EMM2 (Edinburgh minimal medium; Moreno et al., 1991) lacking leucine. In our experiments, 50 µg of one library transformed into 7 × 108 cells generated about 40,000 fission yeast transformants on 45 plates of EMM2 in 90-mm dishes.
C. Microscopic Screening of S. pombe Transformants
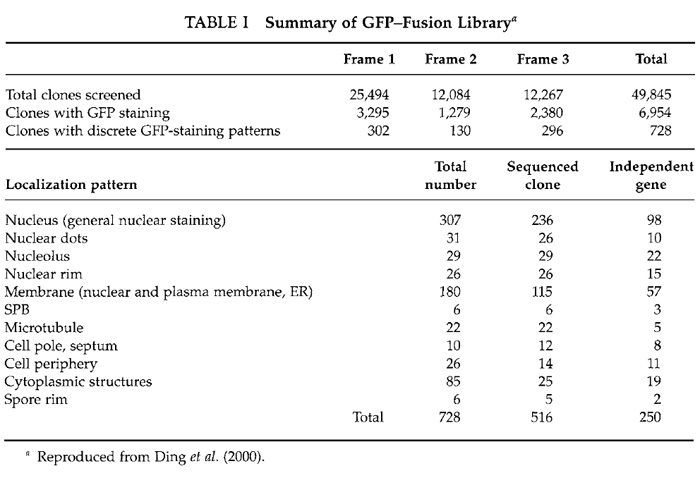
A total of about 50,000 transformants from the three libraries were selected for microscopic screening (Table I). Single colonies were picked with toothpicks and transferred onto new plates of EMM2. Cells were grown for 24h at 33°C and then for 12h at 26°C. Culture of the cells at 26°C enhances GFP fluorescence. Cells were then suspended in liquid EMM2 lacking a nitrogen source on a 10-well glass slide (Polysciences) and were observed with our CCD microscope system using an Olympus oil immersion objective lens (SPlan Apo 60/NA 1.4) and high-selectivity excitation and barrier filters for fluorescence (Chroma Technology, Brattleboro, VT). GFP signals are usually bright and easily detected by eye, but when the signal is weak it is recommended that images be taken on the CCD with relatively long exposure times (a few seconds on our system). Note that dead cells often produce strong autofluorescence at the emission wavelength of GFP and should therefore be avoided when ascertaining the intracellular localization of GFP-fusion proteins.
 |
D. Recovery of Plasmids from Selected Transformants and Determination of Nucleotide Sequences of a DNA Insert
Out of 50,000 transformants, 728 transformants exhibited distinct patterns of GFP fluorescence (Table I). Examples of localizations of GFP-fusion proteins are shown in Fig. 2. Plasmids from these transformants were recovered using standard methods (Moreno et al., 1991). Isolated plasmids were retransformed into fission yeast cells to confirm the intracellular localizations determined from the first screening. Partial nucleotide sequencing of the ORF of the inserted yeast gene was performed using a portion of the GFP gene as a sequencing primer (Fig. 1). Sequencing identified 250 independent genes (Ding et al., 2000).
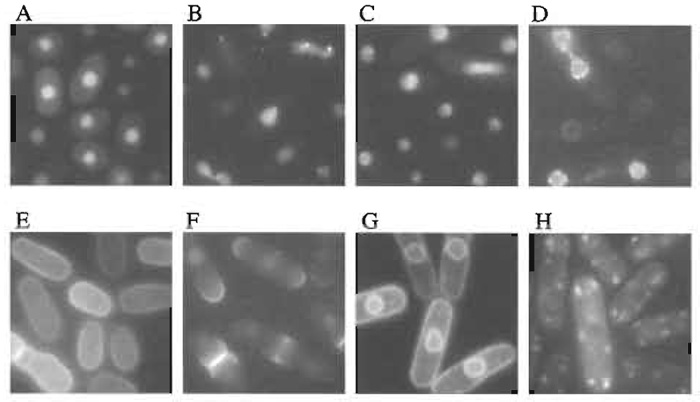 |
| FIGURE 2 Examples of localizations of GFP-fusion proteins. (A) General nuclear staining, (B) nuclear dots, (C) nucleolus, (D) nuclear rim, (E) cell periphery, (F) cell pole and septum, (G) membrane, and (H) cytoplasmic dots. Scale bar: 10 µm. |
E. Construction of Image Database of Intracellular Localization
We constructed an image database of the intracellular localization of these 250 gene products and categorized them into 11 groups as shown in Table I (Ding et al., 2000). Occasionally, a GFP-fusion protein was observed in more than one intracellular localization, as some proteins localize to several structures simultaneously or localize to various different structures during passage through the cell cycle. In these cases, we categorized the gene by the intracellular location that exhibited the most prominent staining. Secondary localizations are also listed in the database and genes can be searched for by any of their detected locations. Using this database, DNA sequences can be searched based on the localization of their products (see our Web site, http://www.karc.crl.go.jp/d332/CellMagic/index.html).
F.Time-Lapse Observation in Living Cells
Time-lapse images of GFP fluorescence in living cells were obtained on a cooled CCD using our computer-controlled fluorescence microscope system (Ding et al., 1998; Haraguchi et al., 1999). For time-lapse observation, living fission yeast cells expressing a GFP-fusion construct are mounted between two coverslips (Method 1) or in a 35-mm glass-bottom culture dish (MatTek Corp., Ashland, MA) coated with concanavalin A (Method 2), and observed in EMM2 medium. Sandwiching between coverslips generates clearer images, but allows observation for only about 2h due to the limited amount of nutrition and oxygen available to the cells in such an arrangement. For longer observation, the use of glass-bottom culture dishes is recommended.
Solution
To make 1 liter of EMM2, add 20g glucose, 5g NH4Cl, 2.2 g Na2HPO4, 3 g potassium hydrogen phthalate, 20ml salts, 1 0.1 ml trace elements, 2 1 ml vitamins, 3 and 20ml amino acids 4.
1 Salts (50x stock): 52.5 g/liter MgCl2·6H2O, 0.735 g/liter CaCl2·2H2O, 50 g/liter KCl, and 2 g/liter Na2SO4.
2 Trace elements (10,000x stock): 5g/liter boric acid (HBBO3), 4 g/liter MnSO4, 4 g/liter ZnSO4·7H2O, 2 g/liter FeCl2·6H2O, 0.4g/liter molybdic acid (H2MoO4), 1g/liter KI, 0.4g/liter CuSO4·5H2O, and 10 g/liter citric acid.
3 Vitamins (1000x stock): 1g/liter pantothenic acid, 10g/liter nicotinic acid, 10 g/liter myo-inositol, and 10mg/liter biotin.
4 Amino acids (50x stock): 10mg/ml L-leucine, 3.75mg/ml adenine sulfate dihydrate, 3.75 mg/ml uracil, 3.75 mg/ml L-histidine hydrochloride monohydrate, and 3.75mg/ml L-lysine hydrochloride.
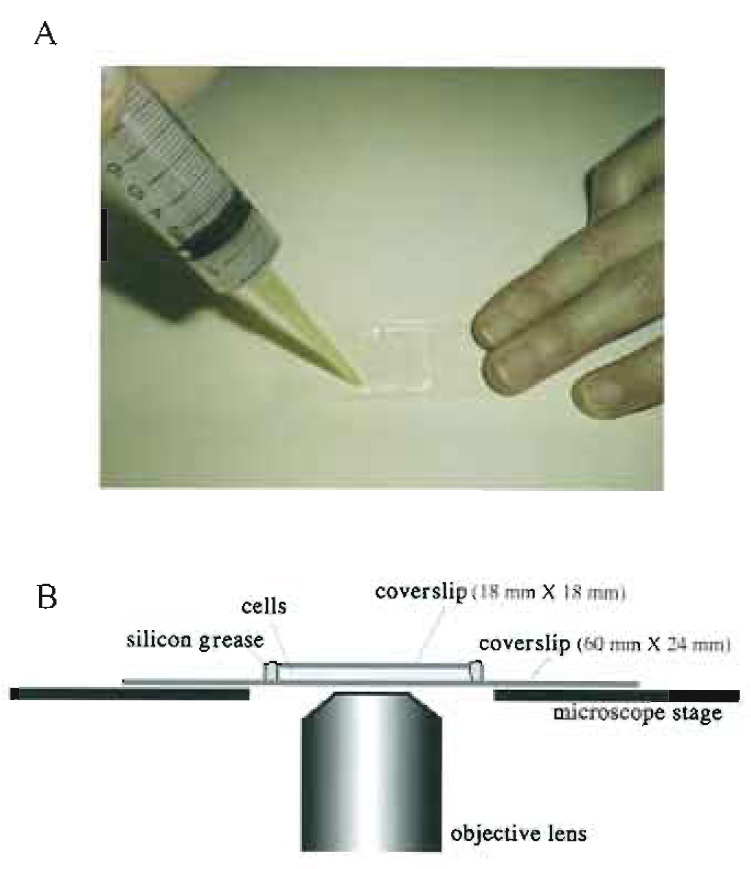 |
| FIGURE 3 Living cells between coverslips. (A) living fission yeast cells are mounted between coverslips and sealed with silicon grease. Silicon grease is packed in a syringe capped with a micropipette tip and applied to the edges of the smaller. upper coverslip. (B) A side view of a specimen on the microscope stage. |
Method 1: Sandwiching between Coverslips
(Fig. 3)
Steps
- Prepare two coverslips (60 x 24mm and 18 x 18mm).
- Suspend fission yeast cells in the EMM2 medium.
- Place 2.5µl of cells on the larger coverslip (60 x 24mm).
- Cover with the smaller coverslip (18 x 18 mm) carefully so as not to introduce air bubbles.
- Seal with silicon grease to avoid evaporation during observation (Fig. 3A). Do not use organic solventcontaining sealing reagents such as nail enamel as they are toxic to living cells. If the sealing is poor, cells can move due to streaming of media caused by evaporation.
- Observe the specimen on an inverted microscope (Fig. 3B).
 |
| FIGURE 4 Living cells in a glass-bottom culture dish. (A) A 35-mm glass-bottom culture dish. (B) A side view of a specimen on the microscope stage. |
Steps
- Coat a glass-bottom culture dish (Fig. 4A) with concanavalin A shortly before use. Spread 50µl of 1 mg / ml concanavalin A solution on the glass bottom of the dish. Then, remove excess solution and dry.
- Suspend fission yeast cells in EMM2 medium.
- Place 50 µl of cells on the glass bottom in the center of the dish. The cell wall will adhere to the concanavalin A.
- Place small drops of water, or small pieces of wet paper or cotton, at the periphery of the dish to avoid evaporation during observation. Seal the lid of the dish with parafilm.
- Place the dish on an inverted microscope (Fig. 4B). Leave the specimen on the microscope stage for about 10min before observation to allow the cells to settle down in the dish.
IV. COMMENTS
A spacer between GFP and the gene may affect the localization of the GFP-fusion protein. We constructed our library using three plasmid vectors, pEG3-1, pEG3-2, and pEG3-3. In pEG3-1, there was no spacer codon between GFP and the yeast coding sequence, whereas in pEG3-2, three amino acid residues, Trp, Gly, and Ser, were inserted between GFP and the yeast coding sequence, and in pEG3-3 three amino acid residues, Leu, Gly, and Ser, were inserted. The best results were obtained when Leu, Gly, and Ser were inserted between GFP and the fission yeast coding sequence (Ding et al., 2000). Fusing the GFP directly to the protein backbone or with a spacer made of a heterocyclic aromatic amino acid, such as tryptophan, may affect the secondary structure of the fusion protein and, consequently, may disturb its intracellular localization.
An obvious limitation of our library is that those gene fragments that abate cell viability cannot be obtained. Another limitation of our library is occasional mislocalization of the fusion gene products. Because we fused GFP to random fragments of the genomic DNA, the C-terminal portion of the gene product was truncated to various extents and replaced by GFP. Gene products that have localization signals at their C termini could be mislocalized or excluded during screening. Also, cryptic localization signals can occasionally be contained within the amino acid sequences fused to GFP. Now that all the proteincoding genes of S. pombe have been identified by the completion of the S. pombe genome project (Wood et al., 2002), these limitations can be partly eliminated by constructing an ordered library in which the GFP gene is fused to full-length open reading frames. In the budding yeast S. cerevisiae, a collection of strains have been constructed, in which the GFP-fusion construct is integrated into the native chromosomal locus and expressed under their own promoter (Huh et al., 2003).
V. PITFALLS
- For observing living fluorescently stained cells, use a high-sensitive camera to avoid damage to the cells caused by the excitation light and also minimize the excitation by the use of a shutter.
- Occasionally an isolated plasmid will contain two different GFP-fusion genes, presumably as a result of heterodimer formation. This is seen during nucleotide sequence determination as two sequences overlapping each other. In such cases, the fusion proteins must be recloned into separate plasmids and the intracellular localization of the GFP-fusion products examined individually.
- Because the GFP-fusion constructs are expressed on a multicopy plasmid, overexpression of the GFP-fusion protein may occur and affect its intracellular localization. To minimize this problem, it is recommended to use fresh preparations of exponentially growing cells for observation. Cells with physiological perturbations tend to fluoresce brightly or to accumulate fluorescent protein aggregates in the cell. Avoid observing such cells that give atypically bright fluorescence. In principle, localization patterns representative of the major population of cells in a microscope field are reliable.
References
Ding, D. Q., Chikashige, Y., Haraguchi, T., and Hiraoka, Y. (1998). Oscillatory nuclear movement in fission yeast meiotic prophase is driven by astral microtubules, as revealed by continuous observation of chromosomes and microtubules in living cells. J. Cell Sci. 111(Pt. 6), 701-712.
Ding, D. Q., Tomita, Y., Yamamoto, A., Chikashige, Y., Haraguchi, T., and Hiraoka, Y. (2000). Large-scale screening of intracellular protein localization in living fission yeast cells by the use of a GFP-fusion genomic DNA library. Genes Cells 5, 169-190.
Haraguchi, T., Ding, D. Q., Yamamoto, A., Kaneda, T., Koujin, T., and Hiraoka, Y. (1999). Multiple-color fluorescence imaging of chromosomes and microtubules in living cells. Cell Struct. Funct. 24, 291-298.
Helm, R., and Tsien, R. Y. (1996). Engineering green fluorescent protein for improved brightness, longer wavelengths and fluorescence resonance energy transfer. Curr. Biol. 6, 178-182.
Huh, W. K., Falvo, J. V., Gerke, L. C., Carroll, A. S., Howson, R. W., Weissman, J. S., and O'Shea, E. K. (2003). Global analysis of protein localization in budding yeast. Nature 425, 686-691.
Matsumoto, T., Fukui, K., Niwa, O., Sugawara, N., Szostak, J. W., and Yanagida, M. (1987). Identification of healed terminal DNA fragments in linear minichromosomes of Schizosaccharomyces pombe. Mol. Cell. Biol. 7, 4424-4430.
Maundrell, K. (1993). Thiamine-repressible expression vectors pREP and pRIP for fission yeast. Gene 123, 127-130.
Moreno, S., Klar, A., and Nurse, R (1991). Molecular genetic analysis of fission yeast Schizosaccharomyces pombe. Methods Enzymol. 194, 795-823.
Sawin, K. E., and Nurse, P. (1996). Identification of fission yeast nuclear markers using random polypeptide fusions with green fluorescent protein. Proc. Natl. Acad. Sci. USA 93, 15146-15151.
Wood, V., et al. (2002). The genome sequence of Schizosaccharomyces pombe. Nature 415, 871-880.
Support our developers




