Labeling of Endocytic Vesicles Using Fluorescent Probes for Fluid-Phase Endocytosis
I. INTRODUCTIONEndocytosis occurs by the invagination of plasma membrane to form an intracellular vesicle. Endocytic vesicles, and the intracellular organelles they communicate with, can be labeled by inclusion of fluorescent, membrane-impermeant molecules in the extracellular medium. Such probes are enclosed in the vesicles as they form and remain contained in endocytic compartments. Ultimately, these fluid-phase endocytic probes either accumulate in lysosomes, where they may be degraded, or are returned to the extracellular medium by recycling vesicles. Fluid-phase endocytosis is also called pinocytosis, which is sometimes subdivided into macropinocytosis and micropinocytosis, the formation of large and small pinosomes, respectively (Maniak, 2001; Swanson, 1989a; Swanson and Watts, 1995). Macropinocytosis is specialized for fluidphase uptake, whereas micropinocytosis involves both receptor-mediated (membrane adsorptive) and fluid-phase uptake. Receptor-mediated endocytosis via clathrin-coated vesicles can be distinguished from fluid-phase endocytosis by using fluorescent specific ligands for the receptor, e.g., FITC-labeled transferrin for the transferrin receptor. Compared with receptormediated endocytosis, the biological function of macropinocytosis has been less investigated so far. However, attention has been increasingly paid to this fluid-phase endocytic pathway (Araki et al., 2000, 2003; Maniak, 2001) because it has been known that macropinocytosis is utilized for antigen presentation in dendritic cells and macrophages (Norbury et al., 1995; Sallusto et al., 1995; Nobes and Marsh, 2000).
A variety of fluorescent probes can be used to label endocytic organelles in living cells. Vital staining techniques using such probes are useful not only for marking endocytic organelles, but also for exploring the kinetics of soluble molecules and the dynamics of endocytic organelles in the cell. This article describes methods for labeling and observing these compartments in living cells and for immunofluorescence microscopy of similarly labeled cells. In addition, it describes a method for the quantitative measurement of fluid-phase pinocytosis using fluorescent probes. These methods are optimized for the study of macrophages, which are actively endocytic cells. It should be kept in mind that most other kinds of cells exhibit lower rates of endocytosis and different kinetics of delivery from endosomes to lysosomes. One may therefore have to extend incubation times or increase probe concentrations to obtain strong fluorescent signals in the microscope or fluorometer. For any new cell type, compartment identities should be determined empirically by comparing fluorescent probe distributions in cells fixed after various pulse-chase intervals with the immunofluorescent localization of known markers for endocytic compartments (e.g., Racoosin and Swanson, 1993).
II. MATERIALS AND INSTRUMENTATION
The fluorescent fluid-phase probes, fluorescein dextran, MW 3000 (FDx3, Cat. No. D-3305), MW 10,000 (FDxl0, Cat. No. D-1821), lysine fixable fluorescein dextran, MW 10,000 (Cat. No. D-1820), and Texas red dextran, MW 10,000 (TRDxl0, Cat. No. D-1828), are from Molecular Probes. Lucifer yellow CH (Cat. No. 86150-2) is from Aldrich. Fluorescein dextran, MW 150,000 (FDx150 Cat. No. FD-150), paraformaldehyde (Cat. No. P-6148), bovine serum albumin (BSA, fraction V., Cat. No. A-9647), Triton X-100 (Cat. No. T- 9284), saponin (Cat. No. S-4521), HEPES (Cat. No. H-3375), and Trizma base (Cat. No. T-1503) are from Sigma Chemical. The 25% glutaraldehyde solution (EM grade, Cat. No. SP-17003-92) is from Nacalai Tesque. A primary antibody, rabbit anti-cathepsin D serum, was a gift from Dr. S. Yokota, Yamanashi Medical School. Monoclonal antibodies recognizing some marker proteins for endocytic compartments are available from the Developmental Studies Hybridoma Bank or commercial sources. Texas red-labeled antirabbit IgG (goat) (Cat. No. TI-1000) is from Vector Lab. Dulbecco's modified essential medium (DMEM, Cat. No. 31600-034), fetal bovine serum (FBS, Cat. No. 16000), and goat serum (Cat. No. 16210) are from GIBCO BRL. Circular glass coverslips 12 and 25 mm in diameter (No. 1 thickness, Cat. No. 12-545-102) and silicon oil (Cat. No. 5159-500) are from Fisher Scientific. Twenty-four-well (Cat. No. 430262) and 6-well (Cat. No. 430343) plates are from Corning Costar. A water aspirator with a Pasteur pipette and a digital micropipette (Nichipet EX1000) are used for quickly replacing media in cell culture wells. The Attofluor cell chamber (Cat. No. A7816) is from Molecular Probes. PermaFluor aqueous mounting medium (Cat. No. 434980) is from Lipshaw/Immunon.
An epifluorescence microscope (Axiophot, Carl Zeiss) is used for observation of both living and fixed cells. A Lucifer yellow filter set is from Omega Optical (Set No. XF-14), in addition to a conventional fluorescein filter set. For high-resolution observation, a 100× PlanApo lens, numerical aperture (NA) 1.4 or a 100× Plan-Neofluar lens, NA 1.3 is used. An inverted-type fluorescence microscope (Nikon TE300) equipped with a cooled CCD camera (Retiga EXi F-M-12-C, QImaging), light path shutter (Uniblitz VMMD1, Vincent Associates), and a thermo-controlled stage (Model TC- 102, Harvard Apparatus) are used for longer observations of living cells. A personal computer (Precision 360, Dell) installed with MetaMorph version 4.6 imaging system software (Universal Imaging Co.) is used for collecting images from the CCD camera and for making time-lapse movies. For quantitation of fluorescent probes, a spectrofluorometer (Hitachi 650-40) is used.
III. PROCEDURES
A. Fluorescence Microscopy of Endocytic Compartments Labeled with Fluorescent Probes
Solutions
- Ringer's buffer + bovine serum albumin (BSA) (RB): 155mM NaCl, 5mM KCl, 2mM CaCl2, 1 mM MgCl2, 2 mM NaH2PO4, 10 mM HEPES, 10 mM D-glucose, pH 7.2, plus 0.05% BSA. To make 1 liter, add 9.1 g of NaCl, 0.37g of KCl, 0.275g of NaH2PO4·H2O, 2.38g of HEPES, 1.8g of D-glucose, 0.22 g of CaCl2, 0.2 g of MgCl2·6H2O, and 0.5g BSA to 950ml distilled water, adjust to pH 7.2 with 1 N NaOH, and bring the volume to 1 liter. Sterilize with a 0.22-µm filter and store at 4°C.
- Fluorescent probe stock solutions: Dissolve 10mg of fluorescent probes such as Lucifer yellow, FDx3, FDx10, FDx150, and TRDxl0 in 1 ml PBS. Lysine fixable dextran probes can be used to increase fluorescent signals in aldehyde-fixed cells. Divide in 200-µl aliquots in tubes and store at -20°C.
- Labeling medium: Dilute fluorescent probe solutions to a final concentration at 1.0mg/ml in RB. A volume of 0.4 ml labeling medium is required for each well of a 24-well dish. The concentrations of probes may be changed depending on cell types. One can differentially label macropinosomes and micropinosomes using different sized probes (Fig. 1). To label both macropinosomes and micropinosomes, use low molecular weight probes such as Lucifer yellow and FDx3. To label primarily macropinosomes, use larger probes, such as FDx150 (Araki et al., 1996). Warm to 37°C before adding to cells.
- 8% paraformaldehyde stock solution: To make 50 ml, add 4 g paraformaldehyde to 30ml distilled water and heat to 70°C while stirring. Add a few drops of 1N NaOH so that the mixture becomes clear. Bring the final volume to 50ml with distilled water and filtrate with paper filter. This solution may be kept in aliquots at -20°C. To make up a fixative, thaw an aliquot in a hot water bath (~50°C) until the solution becomes clear.
- 80mM HEPES stock solution, pH 7.2: To make 100ml, add 1.91g of HEPES to 70ml of distilled water. Adjust pH to 7.2 with 1 N NaOH while stirring. Bring the final volume to 100ml with distilled water. Store at 4°C.
- Fixative: 4% paraformaldehyde and 0.1% glutaraldehyde in 40mM HEPES buffer, pH 7.2, containing 6.8% sucrose. To make 10ml, add 5ml of 8% paraformaldehyde solution, 40µl of 25% glutaraldehyde solution, and 0.68g of sucrose to 5 ml of 80mM HEPES, pH 7.2.
- Phosphate-buffered saline (PBS): To make 5 liters, dissolve 40 g of NaCl, 1 g of KCl, 7.1 g of Na2HPO4, and 1 g of KH2PO4 in 4 liters of distilled water. Bring the final volume to 5 liters with distilled water. The solution should be pH 7.2.
Steps
- Culture cells on 12-mm, circular, No. 1 thickness coverslips in 24-well culture dishes in DMEM with 10% heat-inactivated FBS.
- Aspirate the culture medium from the cell culture well and add prewarmed labeling medium. Swirl the dishes and incubate cells in labeling medium for various times at 37°C. Then rinse quickly by changing warm RB to remove fluorescent probe and chase in RB as necessary. For mouse macrophages, a 2- to 5- min labeling incubation without chase will label early endosomes, including micro- and macropinosomes (Fig. 1). A subsequent chase in the absence of the probe for 5 to 15 min should label late endosomes. A 30- to 60-min labeling incubation labels all endocytic compartments, including early and late endosomes and lysosomes, and a 30-min labeling followed by a chase longer than 60min should label primarily lysosomes (Fig. 2).
- Fix the cells in 4% paraformaldehyde and 0.1% glutaraldehyde in 40mM HEPES, pH 7.2, containing 6.8% sucrose for 30 to 60min at 37°C.
- Rinse 3 × 5 min with PBS.
- Mount the coverslip, cell side down, on a slide using mounting medium. Seal with nail polish between the coverslip and the slide.
- Observe the slide with an epifluorescence microscope. We can observe living cells without fixation for short periods using a simple microscope culture chamber. This method has been described in detail previously (Swanson, 1989b; Raccosin and Swanson, 1994). Briefly, assemble a chamber on a slide using small coverslip fragments to support the coverslip. RB should be added to fill the space between the slide and the coverslip. Seal the coverslip to the slide using a heat-melted paraffin-based compound (Swanson, 1989b). The method for longer observations of living cells is described next.
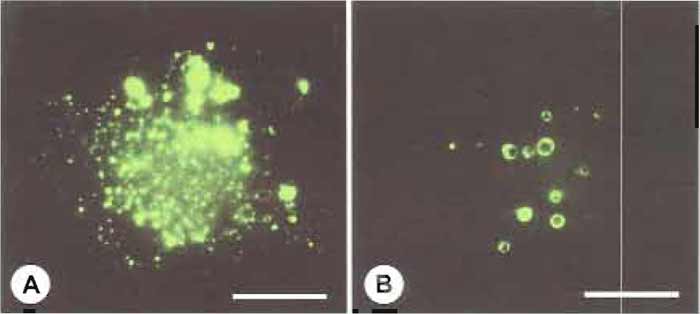 |
| FIGURE 1 Differential labeling of macropinosomes and micropinosomes with Lucifer yellow (A) and
FDx150 (B). Macrophages were incubated for 5 min in labeling medium containing Lucifer yellow or FDx150,
washed briefly, and fixed immediately. A low molecular weight probe, Lucifer yellow (MW 457), labels both
macropinosomes and micropinosomes (A). A larger probe, FDx150 (MW 150,000), labels predominantly
macropinosomes (B). Bars: 10µm. |
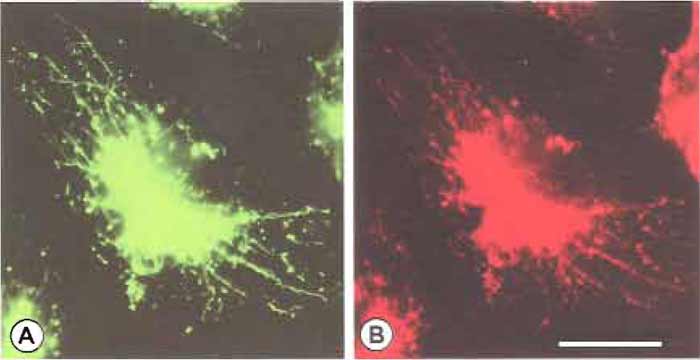 |
| FIGURE 2 Immunofluorescence of cathepsin D in endocytic compartments labeled with endocytic probes. Macrophages were incubated for 30min in labeling medium containing 0.5mg/ml lysine-fixable FDxl0 and were chased for 30min. Cells were then fixed and processed for immunofluorescence using a primary antibody against cathepsin D and the Texas red-conjugated secondary antibody. (A) Fluorescein image shows that FDxl0 labels tubular lysosomes. (B) Texas red image shows that tubular lysosomes, which are labeled with FDxl0, are positive for cathepsin D. Bar: 10µm. |
B. Observation of Endocytic Compartments in Living Cells
Step
- Plate cells on 25-mm No. 1 coverslips in a 6-well culture dish containing tissue culture medium.
- Replace the culture medium with labeling medium. Incubate in labeling medium for various times at 37°C as described earlier, to label the endocytic compartments.
- Wash away fluorescent probe from the coverslip with RB.
- Assemble the coverslip in an Attofluor chamber and fill with 1 ml of RB. You may slowly add a small amount of silicon oil to cover the surface of RB in the chamber. The thin layer of silicon oil protects evaporation of RB during long observations.
- Put one drop of immersion oil on the objective lens of an inverted microscope and settle the Attofluor chamber in the Leiden chamber thermocontrolled at 37°C on the microscope stage.
- Observe the coverslip and acquire images using a high sensitive cooled CCD camera under the lowest light exposure as possible, as intense excitation light may cause not only photobleaching, but also photochemical damage to living cells. Under optimal conditions of labeling, we can observe cells for several minutes under conditions of low-intensity illumination. This can be extended to an hour by inserting a shutter into the light path to control exposures.
- Collect time-lapse images using MetaMorph imaging software, which controls the CCD camera and the light path shutter. These collected images can be saved as a stack file in electronic storage media such as CD-R or DVD-RAM for later processing into movies of living cells. For further information of imaging techniques, see also the sections on digital video microscopy and fluorescent microscopy of living cells in Volume 3.
C. lmmunocytochemical Characterization of Endocytic Compartments Labeled with a Fluorescent Probe
Solutions
- Fixative: Prepare 10ml of 4% paraformaldehyde in 40mM HEPES buffer, pH 7.2, containing 6.8% sucrose
- 0.25% NH4Cl in PBS (NH4Cl/PBS): To make 100ml, dissolve 0.25 g of NH4Cl in 100 ml PBS
- 0.25% saponin or Triton X-100, 2% BSA in PBS (permeabilizing/ blocking buffer): To make 50ml, dissolve 1 g BSA and 125 mg saponin or Triton X-100 in 50 ml of PBS
- Primary antibody: Dilute serum or antibody with permeabilizing/blocking buffer. In the example shown, we diluted rabbit anti-cathepsin D serum at 1:500
- Secondary antibody: Dilute Texas red-labeled antirabbit IgG with PBS at 1:250-500
Steps
- Incubate the cells with labeling medium as described earlier. Lysine-fixable FDx or Lucifer yellow should be used when the other markers are to be localized by immunofluorescence. Nonfixable FDx would be lost during permeabilization of the cell.
- Rinse in PBS to remove excess fluorescent probe, unless the cells were chased in RB without fluorescent probe.
- Fix in 4% paraformaldehyde in 40mM HEPES buffer, pH 7.2, containing 6.8% sucrose for 30-60min at 37°C. Glutaraldehyde may be added to the fixative (0.1%), providing that antigenicity is resistant to glutaraldehyde.
- Rinse with PBS 3 × 5 min and further immerse in NH4Cl/PBS for 10min to quench free aldehyde.
- Treat cells with permeabilizing/blocking buffer for 2 × 5 min.
- Put parafilm in a container with a moist paper. Place one 40-µl drop of primary antibody on the parafilm for each coverslip.
- Wipe the cell-free side of the coverslip with Kimwipe paper. Place the coverslip cell side down on a drop of primary antibody. Incubate with the primary antibody for 1 h at room temperature in the moisture chamber. You may prolong the incubation time to overnight at 4°C.
- After incubation, put the coverslip back into the well and wash three times for 5 min each with PBS.
- Using the same method as for the primary antibody, incubate with secondary antibody, e.g., Texas red-conjugated anti-rabbit IgG diluted in PBS at a concentration 1:500, for 1 h at room temperature.
- Wash the coverslip with PBS three times for 5 min each.
- Mount the coverslip on a slide using the mounting medium. Seal the coverslip with nail polish. Observe the specimens with an epifluorescence microscope using fluorescein and Texas red filter sets.
D. Quantitative Fluorometric Analysis of Endocytic Compartments Labeled with Fluorescent Probes
Solutions
- Lysis buffer: 0.1% Triton X-100 in 50mM Tris, pH 8.5. To make 100ml, dissolve 0.6 g of Trizma base and 0.1 g of Triton X-100 in 80 ml of distilled water. Adjust pH to 8.5 with 1 N NaOH. Bring the volume to 100ml.
- PBS: Make 3 liters of PBS as described earlier. Refrigerate before use.
- 0.1% BSA/PBS: To make 2 liters, dissolve 2g of BSA in 2 liters of PBS.
- Standard solutions of fluorescent probes: Dilute the labeling medium to concentrations of 0, 1, 5, 10, and 20ng probe/ml in lysis buffer. Each solution should be more than 2 ml.
Steps
- Plate the cells at a high density (e.g., 2 × 105 cells/well) in a 24-well culture dish. Triplicate experiments are desirable.
- Replace the culture medium with labeling medium containing fluorescent probes. Dual labeling with FDx and Lucifer yellow is possible (Berthiaume et al., 1995). Incubate at 37°C for various times. A 0-min incubation should be done as a control to determine the background level.
- Discard the labeling medium and rinse the culture dish twice by dipping into a l-liter beaker filled with ice-cold 0.1% BSA/PBS for 5min each. Repeat with another beaker filled with cold PBS for 5 min.
- Drain PBS and aspirate remaining PBS completely.
- Put 0.5 ml of lysis buffer into each well and leave it at least 30min to complete cell lysis.
- Fill disposable 1-cm plastic cuvettes with 0.75 ml of lysis buffer. Add 0.4 ml of cell lysate into the plastic cuvette and dilute it with another 0.75 ml of lysis buffer so that the final volume is 1.9ml.
- Measure the fluorescence of lysate in a spectrofluorometer. Fluorescein can be measured at excitation (exc.) 495nm and emission (em.) 514nm. Lucifer yellow is exc. 430nm and em. 580nm. These wavelengths allow selective measurement of FDx and Lucifer yellow when the cells are labeled with both probes. Lucifer yellow alone is best measured at exc. 430 nm and em. 540 nm.
- Measure the protein concentration of lysates remaining in wells using a BCA protein assay kit (Pierce Chemical Co.).
- Prepare the standard solutions of 0, 1, 5, 10, and 20ng probe/ml in lysis buffer and measure in a spectrofluorometer to obtain a standard curve.
- Calculate the amount of probes from the standard curve and express the value as nanogram probe per milligram protein.
IV. PITFALLS
- A prolonged exposure to intense excitation light may cause a release of fluorescent probes from endocytic vesicles into cytoplasm, especially in living cells. To avoid this, reduce the intensity of the excitation light or the exposure time as much as possible (Video 1 and 2 in the online version).
- Lucifer yellow can be seen using some fluorescein filter sets with a wide band pass (e.g., Olympus BP490), but not some others (e.g., Zeiss No. 09). Choose an appropriate filter set for Lucifer yellow (e.g., Omega Optical Set No. XF-14, Zeiss No. 05).
- Many kinds of fluorescent-conjugated probes are available commercially; however, some are not suitable for fluid-phase probes. Texas red albumin is taken up very efficiently by adsorptive endocytosis, although it is sometimes used as a fluid-phase probe. Lysine-fixable Texas red dextran may bind nonspecifically to coverslips and create a high background fluorescence.
- In the combination of fluorescent fluid-phase probe labeling with immunocytochemistry, we are often faced with a problem that fluid-phase probes release from macropinosomes during membrane permeabilization that is necessary for antibody impregnation. In such a case, the author recommends using a mild detergent such as saponin rather than Triton X- 100. The addition of 2% BSA to permeabilizing buffer considerably protects the fluid-phase probe release. The addition of 0.1% glutaraldehyde to the fixative also effectively fixes fluorescent probes in macropinosomes; however, because some antigens lose their antigenicity, you have to examine the resistance of the antigen to glutaraldehyde.
- For quantitative analysis using a less sensitive fluorescence plate reader, plate the cells at a higher density to increase the sensitivity for fluorescence.
References
Araki, N., Hatae, T., Furukawa, A., and Swanson, J. A. (2003). Phosphoinositide-3-kinase-independent contractile activities associated with Fc y-receptor-mediated phagocytosis and macropinocytosis in macrophages. J. Cell Sci. 116, 247-347.
Araki, N., Hatae, T., Yamada, T., and Hirohashi, S. (2000). Actinin-4 is preferentially involved in circular ruffling and macropinocytosis in mouse macrophages: Analysis by fluorescence ratio imaging. J. Cell Sci. 113, 3329-3340.
Araki, N., Johnson, M. T., and Swanson, J. A. (1996). A role for phosphoinositide 3-kinase in the completion of macropinocytosis and phagocytosis by macrophages. J. Cell Biol. 135, 1249-1260.
Berthiaume, E. P., Mediana, C., and Swanson, J. A. (1995). Molecular size-fractionation during endocytosis in macrophages. J. Cell Biol. 129, 989-998.
Maniak, M. (2001). Macropinocytosis. In "Endocytosis" (M. Marsh, ed.), pp. 78-93. Oxford Univ. Press, Oxford.
Nobes, C., and Marsh, M. (2000). Dendritic cells: New roles for Cdc42 and Rac in antigen uptake? Curr. Biol. 10, R739-R741.
Norbury, C. C., Hewlett, L. J., Prescott, A. R., Shastri, N., and Watts, C. (1995). Class I MHC presentation of exogenous soluble antigen via macropinocytosis in bone marrow macrophages. Immunity 3, 783-791.
Racoosin, E. L., and Swanson J. A. (1993). Macropinosome maturation and fusion with tubular lysosomes in macrophages. J. Cell Biol. 121, 1011-1020.
Racoosin, E. L., and Swanson, J. A. (1994). Labeling of endocytic vesicles using fluorescent probes for fluid-phase endocytosis. In "Cell Biology: A Laboratory Handbook" (J. E. Celis, ed.), pp. 375-380. Academic Press, San Diego.
Sallusto, E, Cella, M., Danieli, C., and Lanzavecchia, A. (1995). Dendritic cells use macropinocytosis and the mannose receptor to concentrate macromolecules in the major histocompatibility complex class II compartment: Downregulation by cytokines and bacterial products. J. Exp. Med. 182, 389-400.
Swanson, J. A. (1989a). Phorbol esters stimulate macropinocytosis and solute flow through macrophages. J. Cell Sci. 94, 135- 142.
Swanson, J. A. (1989b). Fluorescent labeling of endocytic compartments. In "Methods in Cell Biology" (Y.-1. Wang and D. L. Taylor, eds.), Vol. 29, pp. 137-151. Academic Press, New York.
Swanson, J. A., and Watts, C. (1995). Macropinocytosis. Trends Cell Biol. 5, 424-428.
Support our developers




