Preparation of Tubulin from Porcine Brain
INTRODUCTORY COMMENTIn the previous edition to this series (2nd Ed., Vol. 2, pp. 205-212, 1998), we described a method to purify tubulin from bovine brains. Since that time there have been concerns in many countries over the risks of handling bovine nervous tissue relating to bovine spongioform encephalitis and its possible connection with human varient Creuzfeldt-Jakob disease. As a result, abattoirs have been reluctant or have even refused to handle these tissues. Consequently we have changed the species source from which we make the tubulin and have amended the title accordingly. However the protocols described are applicable in their entirety to the bovine source. The bovine and porcine equivalents are indicated in the protocol.
I. INTRODUCTION
Recent research on microtubules has given much insight into many fundamental problems in cell biology, such as cell structure and polarity, vesicular transport, cell division, and chromosomal segregation.
Furthermore, the ability to prepare polarity marked microtubules (Howard and Hyman, (1993) has contributed much to our understanding in the behavior of microtubule motors.
Microtubules are polymers of the heterodimeric protein tubulin. Because these heterodimers have the same orientation in the microtubule lattice and the dimers themselves are not symmetric, it follows that the microtubule itself must have an inherent polarity. Polymerization can take place from either end of the growing microtubule, although at different rates, having a slow growing (-) and more rapidly growing (+) end. The tubulin heterodimer has binding sites for two GTP molecules in its unpolymerized state, only one of which (that GTP that is bound to the E site of the tubulin β subunit) is hydrolyzable. While it is not within the scope of this article to examine all the properties of microtubules and tubulin, some of these are of direct relevance to their isolation. The current model first proposed by Mitchison and Kirschner (1984a) and developed further since then (reviewed by Hyman and Karsenti [1996]) suggests that GTP-unpolymerized tubulin is incorporated at the + growing end of the microtubule and that after a delay, during which time more GTP-bound tubulin is bound at the growing end, the GTP on the β subunit is then hydrolysed to GDP, possibly destabilizing the microtubule lattice. Because GTP-bound tubulin dissociates from the microtubules slowly and the dissociation rate of exposed bound GDP tubulin is two or three orders of magnitude higher, it follows that the presence of a socalled GTP-tubulin "cap" will protect the polymerized microtubule from depolymerization. Alternatively, the GTP "cap" structure may be inherently more stable, effectively preventing the GDP (destabilized) polymerized tubulin from dissociating from the lattice. Thus the "GTP cap" hypothesis states that if the microtubule loses this GTP "cap" structure because of slow growth (e.g., due to lack of free tubulin, cold temperature, presence of Ca2+), the GDP-tubulin dissociates from the lattice. Microtubules growing more slowly are more likely to have GDP-tubulin exposed and thus dissociate rapidly, causing microtubule shrinkage.
The preparation of tubulin described here utilizes these phenomena by a series of alternate depolymerizing and polymerizing steps (see Fig. 1) to obtain tubulin and microtubule-associated proteins (MAPs), followed by ion-exchange chromatography to separate the tubulin from MAPs. This protocol is based on that originally described by Shelanski et al. (1973) and modified by Weingarten et al. (1974) and subsequently by Mitchison and Kirschner (1984b). Tubulin can be modified further for other uses (Hyman et al., 1991).
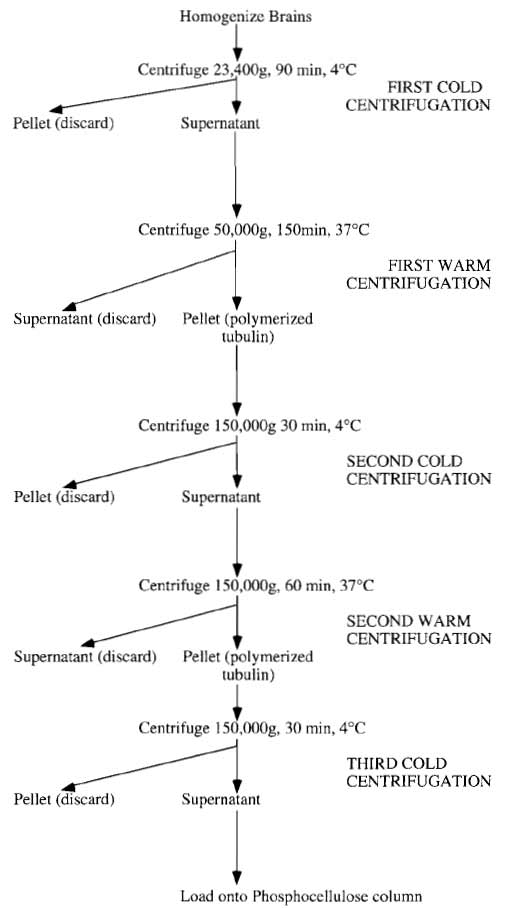 |
| FIGURE 1 Summary of tubulin preparation. |
This article describes how to make pure tubulin with emphasis on large-scale (gram quantities) purifications. This, as will be made clear in later sections, is quite an exercise in both organization and logistics. However, the method can easily be scaled downwards if large amounts of tubulin are not required.
Because the principle of tubulin preparation relies on successive rounds of polymerisation and depolymerisation steps based on temperature shifts, delays and not having the right equipment at the right temperature will reduce the yield drastically. Consequently, after one centrifuge run is completed, it is good practice to reset the temperature for the next spin and switch on the vacuum, checking the centrifuge regularly for faults. This also applies to the rotors, and it is a good idea to have some large water baths set at 37°C and some ice troughs in which to either raise or lower the rotor temperature accordingly. For the polymerization of tubulin, a good supply of hot running water is essential, as slow polymerization will reduce the overall yield.
II. MATERIALS AND INSTRUMENTATION
The protocol, as stated earlier, is suitable for preparation of large (2 to 3 gram) quantities of pure tubulin. To obtain this quantity, it will be necessary to obtain approximately 30 porcine (or 12 bovine) brains. It follows from this that the volumes involved in processing this number of brains, at least in the initial stages of preparation, will be quite large and many centrifuges will be necessary.
A. Centrifuges, Rotors, and Bottles
It is desirable, although not absolutely necessary, to have a centrifuge capable of spinning, at low speed, up to 10 litres of homogenate.
At least six Sorval RC5s, evolutions, or equivalent centrifuge series with SuperLite SLA 1500 are needed together with 36 GSA rotor bottles (250-ml capacity) and their adaptors. The Beckman equivalent to this is the Beckman Avanti using the JLA-16.250 rotor.
Four ultracentrifuges with three Beckman type 19 rotors and 24 corresponding bottles are needed along with four Beckman type 45Ti rotors and 24 (70-ml capacity) polycarbonate bottles.
As can be seen from this list, the rotors required are quite large and heavy and it is therefore important to check the vacuum and refrigeration efficiency of all the centrifuges beforehand using these rotors. Regarding the rotors themselves, these should be inspected beforehand, and any seals and "O" rings should be checked for signs of perishing, replacing where necessary. It is also important that these "O" rings are greased lightly with vacuum grease and that the screw threads for the rotor lids are smeared lightly with Beckman "Spinkote" to ensure that the samples are sealed and that the vacuum is maintained during the run. Finally, the centrifuge rotors should be checked for the condition of the overspeed decals to prevent premature termination of the run. This is particularly true of the Beckman type 19 rotors, which seem especially sensitive on this point.
The centrifuge bottles too should be inspected for cracks and warping (particularly true of the polycarbonate Beckman type 45 bottles), and the lids and caps should be checked for condition and integrity of the "O" ring.
B. Other Equipment
A tissue blender, such as that made by Waring, along with a 4.5-1itre capacity homogenizing beaker and a (motor-driven) continuous flow homogenizer, such as the Yamato LH-22 model for resuspending the tubulin pellets (or a very large Dounce hand homogenizer), as well as a 1-Litre capacity phosphocellulose (PC) column, are needed. Details of how to prepare such a column are described in detail later.
C. Chemicals
All chemicals should be of analytical grade and can be obtained from a variety of suppliers. Chemicals for the following solutions are obtained from the following suppliers.
MES (M-5057), PIPES (P-6757), EGTA (E-4378), MgCl2 (104-20), GTP (G-8877), ATP (A-7699), and β- mercaptoethanol (M-6250) are from Sigma-Aldrich.
EDTA (1.08418.1000), anhydrous glycerol (1.04093. 2500), NaCl (1.06404.1000), NaOH (1.06498.1000), HCl (1.00319.2500), KH2PO4 (4873.1000), and K2HPO4 (5099. 1000) are from Merck.
III. PROCEDURES
A. Preparation of Tubulin
The buffer systems of choice for the preparation of tubulin are 2-[N-MES for the blending buffer (BB) and PIPES for the remainder. When making up K.PIPES buffer solutions, it is first necessary to raise the pH of the solution to about pH 6.0 using KOH in order to get the PIPES to go into solution. This initial pH adjustment must be done without the use of a pH meter (i.e., using pH indicator paper), as any undissolved PIPES can damage the electrode.
Approximately 6 litres of BB is needed. Blending buffer is 0.1M MES, pH 6.5, 0.1 mM EDTA, 2mM EGTA, and 0.5 MgCl2. For 1 litre, use 21.7 g MES, 4ml 0.5M EDTA stock solution, 0.1 ml 0.5M EGTA stock solution, and 0.5 ml 1 M MgCl2 stock solution.
About six litres of polymerization buffer (PB) is needed for a large tubulin preparation; if preferred, it can be made as a 5× stock solution and then diluted with water at 4°C before use. Polymerization buffer is 0.1M PIPES, pH 6.8, 0.5 mM MgCl2, 2.0 mM EGTA, and 0.5 mM EDTA.
Approximately five litres of Column buffer (CB) is needed for such a preparation, which is made up as a 10× solution and diluted with water at 4°C prior to use. Column buffer is 50mM PIPES, pH 6.8, 1 mM EGTA, and 0.2mM MgCl2. To make 1 litre of 10× column buffer, weigh out 151.2 g of PIPES, 3.8g of EGTA, and 2 ms of a 1M stock solution of magnesium chloride and add 45.6g potassium hydroxide. This will adjust the pH to about pH 6.7 and further adjustments can be made using a 10M solution of potassium hydroxide.
The BRB80 conversion buffer is added to the purified tubulin that has been eluted from the phosphocellulose column to convert the buffer from column buffer composition to BRB80 prior to storage of the tubulin. The BRB80 conversion buffer is added in a ratio of I part conversion buffer to 20 parts tubulin in column buffer.
To make up 150mls of BRB80 conversion buffer combine 47.6g PIPES, 4.2ml MgCl2 (1M), and 1.25ml EGTA (0.2M). This should then be brought to pH 6.8 with potassium hydroxide. Having the correct pH is very important.
Nucleotides are made as stock solutions as 100mM ATP and 200mM GTP. It is important that the pH of these nucleotides does not become acidic, as hydrolysis will result. Consequently, they are made up in water and adjusted to pH 7.5 with sodium hydroxide. The ATP solution is made 200mM with respect to MgCl2. This is not the case with the GTP solution, as this will cause precipitation. These solutions should be stored at -80°C until needed.
Protein concentration determinations are made using the Bio-Rad version of the Bradford protein assay, reading the absorption at 595 nm using bovine serum albumin as a reference standard.
Steps
Note: All g forces quoted are Rmax values.
Because brain tissue has a high density of microtubules in the dendrites and axons of its nerve cells, this is the tissue of choice for a tubulin preparation. Porcine brain is readily available from slaughterhouses. It is essential, however, to get the brains as fresh as possible, as protein degradation will begin to take place soon after death. The brains should be warm when they are received and should be plunged immediately into a mixture of ice and saline solution for transport back to the laboratory. Do not use cold brains.
1. Preparation of Brain Tissue for the First Cold Spin
Cautionary note: As alluded to earlier, possible contact with pathogens associated with nervous tissue should be minimized by always handling the brains with gloves. Waste tissue should be disposed of by incineration.
At the laboratory cold room the brains should be stripped of brain stems, blood clots, and meninges (kitchen paper tissue is very good for this purpose). The brains are then weighed, transferred to the Waring blender, and cold BB containing 0.1% β- mercaptoethanol and 1 mM ATP, but without any protease inhibitors, is added in the ratio of 1 litre of buffer per kilogram of brain tissue. The brains are then homogenized twice for approximately 30s each. Typically it can be expected to need approximately 5 litres of BB for 30 porcine (approximately 12 bovine) brains giving about 10 litres of total homogenate. If a large capacity rotor is available, this homogenate can be poured directly into the 1 litre centrifuge bottles and then centrifuged at 12,000g for 15min. Using this option has the advantage that any unhomogenized tissue and air (from the blending procedure) are removed rapidly. The homogenate is centrifuged further at 34,200g for 60min (SLA1500) at 4°C.
2. First Warm Centrifugation
- Pool and pour the supernatants (approximately 5 litres for a 30 porcine brain preparation) into two 5- litre Ehrlenmeyer flasks.
- Add a half volume of anhydrous glycerol that has been prewarmed to 37°C and then adjust the GTP, ATP, and MgCl2 concentrations to 0.5, 1.5, and 3 mM, respectively. It is essential to bring the tubulin solution above 30°C as quickly as possible.
- Warm the tubulin to 30°C under a continuously flowing hot tap while swirling the flask. At some point after the tubulin has reached 30°C it will be noticed that the solution becomes noticeably more viscous, indicating that polymerization is beginning to take place.
- Incubate the tubulin for a further 60min at 37°C in a water bath before recovery by centrifugation at 50,000 g for 150 min at 37°C in Beckman type 19 rotors.
3. Second Cold Centrifugation
- Resuspend the pellets in approximately 700 ml of PB (with 0.1% β-mercaptoethanol) at 4°C. using either a Dounce or a continuous flow homogenizer. At this point, determine the protein concentration of the resuspended pellet solution and lower the concentration to 25mg/ml by the addition of cold PB.
- Leave the solution on ice for 40min to allow for complete depolymerization before centrifuging at 150,000g for 30min in Beckman type 45Ti rotors at 4°C.
4. Second Warm Centrifugation
- Pool the supernatants from this centrifugation and adjust the concentrations of ATP, GTP, and MgCl2 to 1, 0.5, and 4 mM, respectively.
- As before, add a half volume of anhydrous glycerol prewarmed to 37°C and warm the entire mixture to 37°C. Allow the tubulin to polymerize at this temperature for a further 40min prior to centrifugation at 150,000g at 37°C for 1 h in Beckman type 45Ti rotors. If time is short, the polymerized tubulin pellets can be frozen in liquid nitrogen and stored at -80°C.
5. Third Cold Centrifugation
- Collect and resuspend the polymerized tubulin pellets in a total volume of 200ml of PB with 0.1% β-mercaptoethanol using either the continuous flow or the Dounce homogenizer.
- Determine the protein concentration and dilute the solution with PB as necessary to a final concentration of 35 mg/ml.
- Allow the tubulin to depolymerize on ice for a further 40min and then centrifuge at 150,000g for 30 min in the Beckman type 45Ti rotor.
- The supernatant is now ready for loading onto the phosphocellulose column to separate the tubulin from microtubule-associated proteins.
6. Purification of Tubulin over a Phosphocellulose Column
- Load the depolymerised tubulin onto a 1-1itresized column (at 4°C) that has been equilibrated previously in CB. Tubulin should be loaded onto the column at a flow rate of approximately 1.5ml/min. Once loaded, however, the flow rate can be increased to 6ml/min or even faster if the phosphocellulose shows little sign of compressing, although some compression is inevitable.
- The purified tubulin elutes in the flow through. Pool the protein peak and determine the protein concentration. The purified tubulin in CB can then be snap-frozen in liquid nitrogen or converted to tubulin in BRB80 using BRB80 conversion buffer prior to freezing and subsequent storage at -80°C. When run on a 12% polyacrylamide gel, the purified tubulin, when stained with Coomassie, should appear as a single band free of any trace of contaminating proteins. The MAPs are retained on the column and these proteins can be eluted using CB containing 1M NaCl.
B. Cycling Tubulin
It is inevitable that some tubulin will become inactivated or denatured during passage through the phosphocellulose column, as tubulin is an unstable entity. It is therefore recommended to "cycle" the tubulin after it passes through the column. To cycle tubulin means that it is polymerised at 37°C, reisolated by centrifugation through a glycerol cushion, and depolymerised at 4°C prior to freezing. This can be performed either immediately after elution from the column or after it has been stored at -80°C in CB. In both cases the tubulin buffer should be converted to BRB80 as described earlier. There are several advantages to cycling the tubulin: it enriches for active tubulin dimers, removes free nucleotides, removes denatured tubulin and other impurities, and concentrates the tubulin to approximately 10-20mg/ml (cf. 5-10mg/ ml as it comes through the column.). The resulting cycled tubulin is suitable for use in in vitro assays such as video microscopy (e.g., Andersen et al., 1994). It is also recommended to use cycled tubulin for biochemical studies (e.g., purification of MAPs) because it is much easier to control the amount of microtubules formed when this highly active tubulin is used compared to that obtained directly from the phosphocellulose column.
Steps
To obtain a large stock of identical cycled tubulin, it is recommended to cycle approximately 30ml of tubulin obtained directly from the phosphocellulose column.
- Prewarm the rotor (a Beckman Ti50, TLA100, or MLA 80 rotor is suitable) in a water bath at 37°C. Fill the rotor tubes to half their volume with a glycerol cushion consisting of 60% glycerol in BRB80 (no GTP), place these in the rotor, and allow them to equilibrate to 37°C. While this is happening, proceed with polymerisation of the tubulin.
- Thaw the tubulin rapidly using a water bath set at 37°C until the tube is half full of ice and then continue to thaw the remainder on ice. Adjust the solutes to glycerol PB using anhydrous (100%) glycerol. Allow polymerisation to occur for 40min at 37°C.
- Layer the polymerised tubulin onto the prewarmed cushions using tips with large openings to avoid depolymerising the microtubules. Centrifuge at 226,240g (50,000rpm in the Beckman Ti50 rotor) for 60min or, alternatively, at 70,000rpm for 30min in the TLA100 rotor at 37°C.
- Aspirate away the supernatant above the cushion. Rinse the cushion interface twice with water. Aspirate away the cushion. Resuspend the pellet in 0.25x BRB80 + 0.1% β-mercaptoethanol on ice using an homogeniser to depolymerise the microtubules. The volume of buffer used for the resuspension is chosen so that the final tubulin concentration is between 10 and 20mg/ml (based on the assumption that approximately half the tubulin from the phosphocellulose column will polymerise). Incubate on ice for 15min and then add 5x BRB80 to adjust the buffer to 1x BRB80. (Note: The volume of 5x BRB80 to add is 3/16ths of the volume of 0.25x BRB80-tubulin.)
- Sediment the undepolymerised microtubules by centrifuging the sample at 213,483g (70,000rpm in the Beckman TLA100.2 rotor) for 15min at 4°C.
- Aliquots (10-200µl) of the concentrated tubulin can be made, which should then be snap frozen in liquid nitrogen. The tubulin can then be stored either in liquid nitrogen (indefinitely) or at -80°C (for at least 12 months).
C. Preparation of Phosphocellulose Column
For large-scale preparations of tubulin (finally giving 1 g or more of purified tubulin) a phosphocellulose column of approximately 1 litre volume will be necessary. For successful preparation of PC, it has to be equilibrated for short periods first in base and then in acid, interspersed with water washes. This is normally achieved by suspension of the PC in either the acid or the base solution and then rapid filtration over a sintered glass funnel where the PC can be washed with large volumes of water. Large volumes of PC are, however, quite cumbersome to handle and the importance of setting up an efficient filtration system before commencing the PC preparation cannot be overemphasised. With volumes as large as 1 litre it may be unwise to rely on running water aspirators, as the vacuum produced may be insufficient (but for small volumes of PC these may be suitable). It is better to use a membrane-type pump and if, possible, to connect this to a wide, sintered glass funnel over which Whatmann 3 MM filter paper has been placed. Alternatively, and we have used this quite successfully, use some nylon or poiypropylene meshing (available from SpectraMesh), as this allows rapid filtration rates with almost no risk of tear as there is when using Whatmann filter paper.
When considering what volume of phosphocellulose is needed, our laboratory uses 200ml phosphocellulose to obtain 300-600mg of purified tubulin. When equilibrated in buffer, 1 g of PC powder will give between 5 and 6 ml of PC column matrix (depending on the salt concentration), but do allow for some loss due to the removal of fines when calculating the amount of PC to prepare.
Phosphocellulose is obtained from Whatmann Scientific Ltd. and the column and adaptors can be obtained from Kontes, but any column with similar dimensions (10 x 30 x 4.8cm) should be suitable.
Steps
Note: All stages should be performed at 4°C.
- Having determined the amount of phosphocellulose that is needed using the information just given, slowly hydrate the dry powder by washing twice in 95% ethanol, once in 50% ethanol, and finally once in pure water.
- Resuspend the hydrated phosphocellulose in 25 volumes (liquid volume per original dry weight of phosphocellulose) of 0.5 M NaOH and stir gently (stirring too fast produces fines, which will result in slow column flow rates if not removed later) for 5 min. Filter the phosphocellulose rapidly to remove the NaOH solution and continue to rinse with water until the pH of the washings is lower than 10 (usually at least three times the volume of the NaOH solution used).
- As soon as the pH is sufficiently low, resuspend the phosphocellulose in 25 volumes of 0.5M HCl (i.e., the same volume as the NaOH solution as used earlier) and again stir gently for 5min and filter quickly. Continue to wash with water until no longer acid m usually around 10 times the volume of the HCl solution used.
- If the column is not to be poured at this stage, the phosphocellulose should be resuspended and stirred for 5 min in a 2 M solution of potassium phosphate, pH 7.0. The PC can then be resuspended and stored in a solution of 0.5M potassium phosphate, pH 7.0, containing 20 mM sodium azide as preservative. When the phosphocellulose is removed from storage, it should be resuspended and washed in at least 3 volumes of water.
- At this stage, whether the column material was stored or not, the phosphocellulose should be resuspended in at least 3 volumes of water and transferred to a large measuring cylinder. The phosphocellulose should be stirred and then allowed to settle naturally. After the bulk of the phoshocellulose has settled, it will be noticed that there is a cloudy layer just above it. These are the fines and should be removed by aspiration to ensure fast column flow rates. This cycle of resuspension, stirring, and aspiration of the fines should be repeated until no more fines are visible.
- Resuspend the phosphocellulose in column buffer solution and then degas for 30min prior to pouring the column.
- After use, wash the phosphocellulose column extensively and replace the buffer with a 50mM potassium phosphate buffer containing 20 mM sodium azide.
References
Andersen, S., Buendia, B., Domfnguez, J., Sawyer, A., and Karsenti, E. (1994). Effect on microtubule dynamics of XMAP230, a microtubule-associated protein present in Xenopus laevis eggs and dividing cells. J. Cell Biol. 127, 1289-1299.
Howard, J., and Hyman, A. (1993). Preparation of marked microtubules for the assay of microtubule-based motors by fluorescence microscopy. Methods Cell Biol. 39, 105-113.
Hyman, A., Drechsel, D., Kellogg, D., Salser, S., Sawin, K., Steffen, P., Wordeman, L., and Mitchison, T. (1991). Preparation of modified tubulins. Methods Enzymol. 196, 478-485.
Hyman, A., and Karsenti, E. (1996). Morphogenetic properties of microtubules and mitotic spindle assembly. Cell 84, 401-410.
Mitchison, T., and Kirschner, M. (1984a). Dynamic instability of microtubule growth. Nature 312, 237-242.
Mitchison, T., and Kirschner, M. (1984b). Microtubule assembly nucleated by isolated centrosomes. Nature 312, 232-237.
Shelanski, M., Gaskin, E, and Cantor, C. (1973). Microtubule assembly in the absence of added nucleotides. Proc. Natl. Acad. Sci. USA 70(3), 765-768.
Weingarten, M., Suter, M., Littman, D., and Kirschner, M. (1974). Properties of the depolymerisation products of microtubules from mammalian brain. Biochemistry 13, 5529-5537.
Support our developers




