Construction and Propagation of Human Adenovirus Vectors
Adenoviruses (Ads), which have been used extensively as a model system for molecular studies of mammalian cell DNA replication, transcription, and RNA processing, are now being increasingly investigated as potential mammalian expression vectors for gene therapy and for recombinant vaccines (Berkner, 1988; Graham and Prevec, 1992; Hitt et al., 1999). There are many reasons for this renewed popularity of Ad vectors: the 36,000-bp double-stranded DNA genome of Ad is relatively easy to manipulate by recombinant DNA techniques; Ad infects a wide variety of mammalian cell types, both proliferating and quiescent, with high efficiency; the genome does not undergo rearrangement at a high rate; the viral particle is relatively stable; and the virus replicates to high titer in permissive cells, producing up to 10,000 plaqueforming units (PFU) per infected cell. Late in infection, most of the infected cell protein is virally encoded, potentiating the use of replication-proficient recombinant Ads as short-term high-level expression vectors. In nondividing, nonpermissive cells the viral genome may persist as an episome and continue to express for long periods in vitro. This holds true in vivo as well in the absence of an immune response against vectorinfected cells. This article describes methods for inserting foreign genes into the Ad genome and for purifying, growing, and titrating the recombinant viruses. Our vectors are based on the human Ad5 genome, the structure of which is shown in Fig. 1. In a wild-type infection, early genes (Ela, Elb, E2, E3, and E4) are expressed prior to DNA replication, and late gene expression, driven predominantly by the major late promoter at 16 map units, occurs after the initiation of DNA replication. Deletion of the E1 region renders the virus replication defective, which is desirable for most gene therapy applications. However, such vectors must then be propagated in El-complementing cells, such as the 293 cell line (Graham et al., 1977). Deletion of E3, which is nonessential for virus growth in vitro, together with deletion of El, allows insertion of foreign genes up to about 8 kb in length. Without deleting E3, the insertion capacity of Ad is about 5 kb.
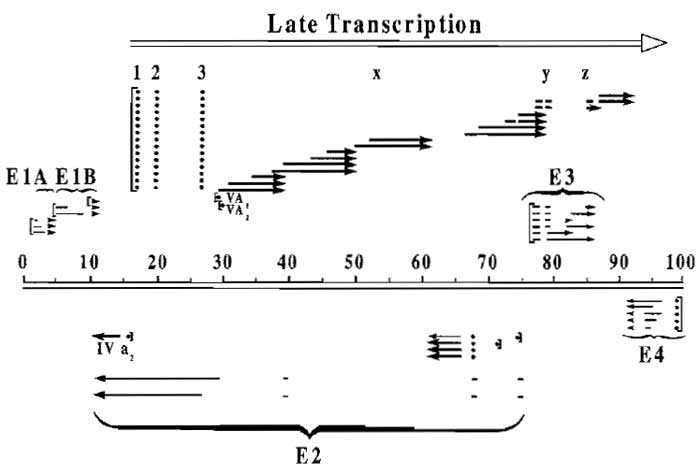 |
| FIGURE 1 Transcription map of the human adenovirus type 5. The approximately 36-kb genome of Ad5 is divided here into 100 map units. Messages from the early regions are indicated as light lines and late messages are indicated in bold. Late transcription originating from the major late promoter at 16 map units and terminating near the right end of the genome is indicated by the open arrow. This transcript is processed into five families of late mRNAs spliced to a common tripartite leader (1, 2, and 3 at map units 16.5, 19.5, and 26.5, respectively), although some mRNA species contain additional leaders. (For more details, see Ginsberg, 1984.) |
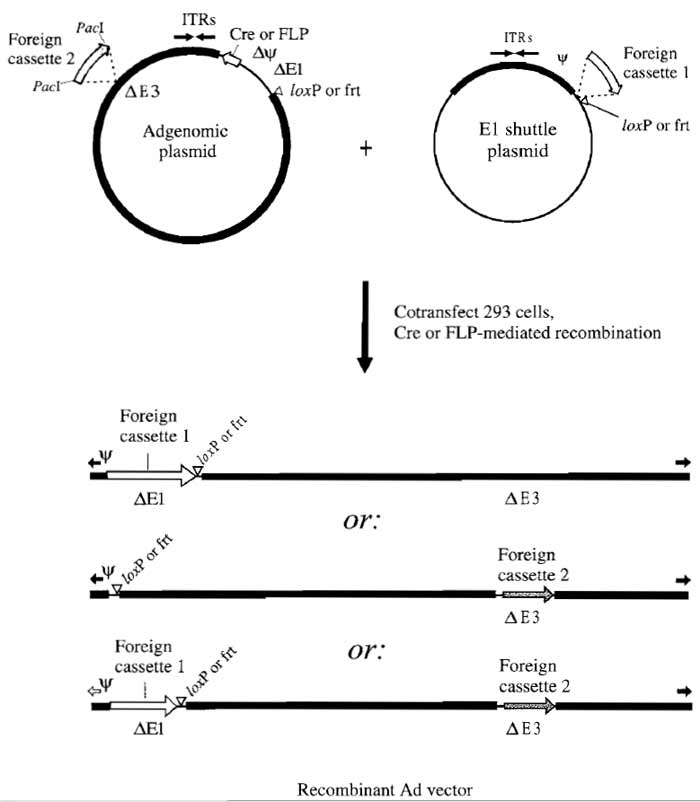 |
| FIGURE 2 Construction of Ad vectors by two-plasmid site-specific recombination in 293 cells. The AdMax strategy used to introduce foreign DNA inserts into the E1 and/or E3 regions for rescue into virus is illustrated. Expression cassettes can be inserted in place of the E1 region (Ad5 nucleotides 455-3523) by cloning into E1 shuttle plasmids carrying the recognition site (e.g., loxP) for a site-specific recombinase. E1 shuttle plasmids are described further in Fig. 3. The 293 cells are then cotransfected with this shuttle plasmid and an Ad genomic plasmid carrying the appropriate recombinase (Cre for loxP-containing shuttles or FLP for frtcontaining shuttles). Genomic plasmids are available with E3 deleted (Ad5 nucleotides 28138-30818; pBHGloxΔE1,E3Cre or pBHGfrtΔE1,E3FLP) as shown or with a wild-type E3 region (pBHGloxE3Cre and pBHGfrtE3FLP) (Ng and Graham, 2002). Expression of recombinase in 293 cells results in recombination between recognition sites in the shuttle and in the genomic plasmid, generating an E1 replacement Ad vector (top vector in illustration). E3 replacement Ad vectors are constructed by inserting the transgene expression cassette into a unique PacI site that replaces the E3 region in the Ad genomic plasmid. The 293 cells cotransfected with this genomic construct and an "empty" (i.e., no transgene) E1 shuttle plasmid will produce a replication-defective E3 replacement Ad vector (middle vector in illustration). Note that it is also possible to cotransfect with a plasmid containing the intact left end of Ad5 to produce an E1+ nondefective vector by overlap recombination (Bett et aI., 1994). Double recombinant vectors are generated by recombination between an E1 shuttle plasmid carrying one expression cassette and a genomic plasmid carrying a second expression cassette (bottom vector in illustration). Ad and bacterial sequences are indicated by thick and thin black bars, respectively, loxP and frt sites by open triangles, inverted terminal repeats (ITRs) by black arrows, and the packaging signal by the symbol Ψ. |
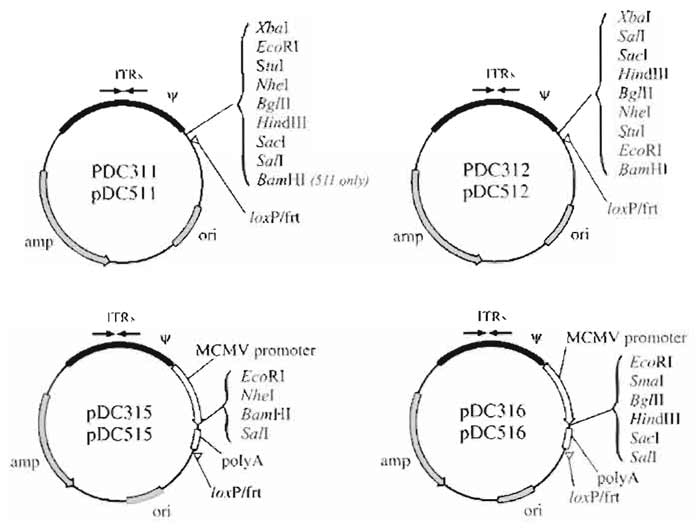 |
| FIGURE 3 Structure of E1 shuttle plasmids used for vector rescue by in vivo site-specific recombination. The shuttle plasmids pDC311, pDC312, pDC315, and pDC316 are used to rescue vectors by Cre-mediated recombination. The shuttle plasmids pDC511, pDC512, pDC515, and pDC516 are used to rescue vectors by FLPmediated recombination. Plasmids pDC311, pDC312, pDC511, and pDC512 are designed for insertion of a cassette consisting of a promoter, transgene, and polyadenylation signal sequence. The polycloning sites of plasmids pDC315, pDC316, pDC515, and pDC516 are flanked 5' by the murine CMV promoter and 3' by the SV40 polyadenylation signal sequence. Coding sequences cloned into the latter plasmids generate vectors with high levels of expression in both human and murine cells (Addison et al., 1997). |
II. MATERIALS AND INSTRUMENTATION
The Cre/loxP and FLP/frt based AdMax vector rescue systems are available, as Kit D (Cat. No. PD-01- 64) and Kit E (Cat. No. PD-01-65) respectively, from Microbix Biosystems, Incorporated. Minimal essential medium (MEM) Fll (Cat. No. 61100-087), L-glutamine (Cat. No. 25030-081), penicillin/streptomycin (Cat. No. 15140-122), horse serum (Cat. No. 16050-159), newborn calf serum (NCS) (Cat. No. 16010-159), agarose (Cat. No. 15510-027), and dithiothreitol (Cat. No. 15508-013) can be obtained from Invitrogen. Bovine serum albumin fraction V (Cat. No. A2153), fetal bovine serum (FBS) (Cat. No. F4135), Joklik's modified MEM (Cat. No. M0518), salmon sperm DNA (Cat. No. D1626), and orcein (Cat. No. 07380) are available from Sigma Chemical Company. All sera are inactivated prior to use by heating to 56°C for 30min. Fungizone can be purchased from Bristol-Myers-Squibb (Cat. No. 043780). Nunc tissue culture dishes (Cat. No. 1- 68381A) can be obtained from VWR. Sterile petri dishes (Cat. No. 08-757-12), Difco agar (Cat. No. 0145- 17-0), Difco Bacto Lennox LB broth base (Cat. No. 0402- 07-0), Becton Dickinson BBL trypticase peptone (Cat. No. Bl1921), and yeast extract (Cat. No. Bl1929) can be obtained from Fisher Scientific. Pronase (Cat. No. 1459643) and bovine pancreatic deoxyribonuclease I (Cat. No. 104-159) can be purchased from Roche Diagnostics. Analytical grade CaCl2·2H2O (Cat. No. B10070) can be obtained from BDH. All other chemicals can be purchased from standard chemical suppliers (e.g., BDH). Spinner flasks (1969 series) can be obtained from Bellco and Pierce Slide-A-Lyzer 10K dialysis cassettes (Cat. No. 66425) from Chromatographic Specialities. Beckman SW41 Ti and SW 50.1 rotors are also required. Reagents for plasmid DNA isolation, as well as restriction enzymes and reagents and apparatus for horizontal slab gel electrophoresis, are described in a number of cloning manuals (e.g., Sambrook and Russell, 2001).
A. Preparation of Plasmid DNA for Cotransfections
In order to minimize the generation of bacterial clones containing rearranged plasmid DNA, which is occasionally observed in preparations of very large plasmids such as pBHGloxΔE1,E3Cre and pBHGfrtΔE1,E3FLP, we have adopted the following protocol for bacterial growth prior to plasmid DNA isolation.
Solutions
- Super broth (SB): Dissolve 5g NaCl, 32 g trypticase peptone, 20 g yeast extract, and 1 g glucose in 1 liter H2O. Add 5 ml 1 N NaOH. Sterilize by autoclaving.
- LB-agar plates: Dissolve 10g BBL Lennox LB broth base in 500ml H2O. Add 7.5g agar and sterilize by autoclaving. Cool LB-agar to about 50°C, add antibiotics as required, and pour 25 ml into each of 20 sterile petri dishes. Store at 4°C.
- Reagents for isolating plasmid DNA on CsCl gradients: Not described here.
Steps
- Streak plasmid-bearing bacteria on an LB-agar plate containing appropriate antibiotics and grow overnight at 37 °C.
- Pick two or more colonies off the plate, resuspend each in 5 ml SB plus antibiotics, and incubate at 37 °C on a shaker for several hours.
- Add each 5ml culture to 500ml SB plus antibiotics and continue incubating overnight.
- Purify the plasmid DNA from each culture separately by alkaline lysis of the bacteria and CsCl banding as described in standard cloning manuals (e.g., Sambrook and Russell, 2001). Plasmid DNA that has not undergone any detectable rearrangement, as indicated from comparison between predicted and observed restriction enzyme cleavage pattens, is suitable for use in cotransfections.
Solutions
- Complete MEMF11 (or Joklik's modified MEM): Add 5 ml 0.2M L-glutamine, 5 ml penicillin/streptomycin (10,000U/ml and 10mg/ml, respectively), and 5ml 0.25mg/ml fungizone to 500ml MEMF11 (or Joklik's modified MEM). Store at 4°C for up to 2 weeks. Add 55 ml heat-inactivated NBS, FBS or HS prior to use in cell culture.
- 10X citric saline: Dissolve 50g KCl and 22 g trisodium citrate dihydrate (Na3C6H507.2H2O) in H2O to a final volume of 500ml. Sterilize by autoclaving and store at 4°C. Dilute 1:10 in sterile H2O to prepare 1X citric saline.
- HEPES-buffered saline (HEBS): Dissolve 5g HEPES free acid, 8g NaCl, 0.37g KCl, 0.1 g Na2HPO4, and 1 g glucose in 900ml H2O. Adjust pH to 7.1. Adjust volume to 1 liter with H2O. Aliquot into small glass bottles, sterilize by autoclaving, and store at 4°C.
- 10X SSC: Dissolve 8.7 g NaCl and 4.4 g tri-sodium citrate dihydrate in H2O to a final volume of 100ml. Adjust pH to 7.0. Autoclave. Prepare 0.1X SSC by diluting 10X SSC and then autoclaving.
- 2mg/ml carrier DNA: Dissolve 100mg salmon sperm DNA in 50ml sterile 0.1X SSC by stirring overnight at room temperature. Determine concentration by reading the OD at 260nm (one OD unit = 50µg/ml). Store in small aliquots at -20°C.
- 2.5M CaCl2: Add H2O to 36.8 g CaCl2.2H2O (analytical grade) to a final volume of 100ml. Sterilize by filtration and store in small plastic tubes at 4°C.
- MEMF11-agarose overlay: To make approximately 200ml, add 10ml horse serum (HS) and 2ml each of Lglutamine, penicillin/streptomycin, fungizone (at concentrations given earlier), and autoclaved 5% yeast extract (w/v in H2O) to 100ml 2X MEMF11. Autoclave 1 g agarose in 100ml H2O. Bring the 2X medium and agarose to 44°C before mixing and use within an hour.
- Grow monolayer cultures of 293 cells in 150-mm dishes in complete MEMFll medium plus 10% FBS (or NBS). At 90% confluence, remove medium, wash each dish twice with 10ml 1X citric saline, and then incubate for a maximum of 15 min at room temperature in 3 ml 1X citric saline to detach cells. Resuspend cells in medium and divide between two or three 150-ram dishes. 293 cells should be refed with fresh medium every 3 days if not ready to passage.
- Set up low-passage (<p40) 293 cells in 60-mm dishes to be about 70-80% confluent at the time of use. As a rule of thumb, one 150-mm dish of nearly confluent 293 cells can be split into eight 60-mm dishes each containing 5ml complete MEMFll + 10% FBS, which will be ready for transfection the next day.
- Add 0.005 volume 2mg/ml carrier DNA to 1X HEBS and shear by vortexing for 1 min.
- For each virus to be rescued, aliquot 2ml HEBS + carrier DNA (enough for four dishes) into each of three sterile clear plastic tubes.
- To these tubes add E1 shuttle plasmid DNA and the appropriate genomic plasmid (e.g., pBHGloxΔE1, E3Cre) in the following amounts: 20µg of each plasmid, 8µg of each plasmid, and 2µg of each plasmid. If a negative control is desired, set up similar coprecipitations omitting the first plasmid. A useful positive control is 2µg of the infectious plasmid pFG140.
- Gently mix by shaking and then slowly add 0.1 ml 2.5M CaCl2 to each tube.
- Gently mix and let stand at room temperature for 15-30 min. (A fine precipitate should form within a few minutes.) Without removing the growth medium, add 0.5ml DNA suspension to each dish of cells (four dishes for each tube of coprecipitate) and then incubate at 37°C in a CO2 incubator for at least 5h, preferably overnight.
- Remove the medium and add to each dish 10ml MEMF11-agarose overlay previously equilibrated to 44°C. After the agarose solidifies, incubate at 37°C. Plaques should appear after about 5-14 days. When dishes are examined from below by eye, plaques appear turbid as a consequence of light scattering by dead cells in an otherwise smooth cell monolayer. Microscopic examination reveals plaques as zones of dead or lysed cells surrounded by rounded infected cells.
- At about 10 days posttransfection, pick wellisolated plaques by punching out agar plugs from the cultures using a sterile Pasteur pipette. Transfer each agar plug to 1 ml sterile PBS++ + 10% glycerol in a sterile vial. Store at -70°C until use.
The following protocol describes the expansion of plaque isolates by growth in monolayer cultures of 293 cells. A portion of the infected cell material is stored for further purification; the remainder is harvested for analysis of viral DNA.
Solutions
- Phosphate-buffered saline++ (PBS++): To make solution A, dissolve 80g NaCl, 2g KCl, 11.5g Na2HPO4, and 2g KH2PO4 in H2O to a final volume of 1 liter. To make solution B, add 1 g CaCl2.2H2O to 100 ml H2O. To make solution C, add 1 g MgCl2.6H2O to 100ml H2O. Sterilize solutions separately by autoclaving. For 100 ml PBS++ mix 88 ml sterile H2O with 10 ml solution A and 1 ml each of solutions B and C.
- PBS++ + 10% glycerol: Add 10 ml sterile glycerol to 90ml PBS++.
- Pronase stock solution: Dissolve 0.5g pronase in 100ml 10mM Tris-HCl, pH 7.5; heat at 56°C for 15 min and then incubate at 37°C for 1 h. Aliquot and store at -20°C To prepare working solution, thaw stock solution just before use and add 0.1 volume to 10mM Tris-HCl, pH 7.5, 10 mM EDTA, 0.5% (w/v) sodium dodecyl sulfate (SDS).
- Complete MEMF11 + 5% HS: Add 25ml HS to 475 ml complete MEMF11.
- 0.1X SSC: See Section III.B, solution 4.
- Reagents for restriction analysis of viral DNA: Not described here.
- Set up 60-mm dishes of 293 cells to be 80-90% confluent at time of infection. The denser and older the cell monolayer, the longer it takes for the cytopathic effect to reach completion.
- Remove medium from 293 dishes and add 0.2 ml virus (agar plug suspension). Rock dishes once and adsorb at room temperature for 30 min. Add 5 ml complete MEMF11 + 5% HS and incubate at 37°C
- A cytopathic effect should be visible within 1-2 days. Harvest virus and extract infected cell DNA (steps 4 to 6) when all cells are rounded and most have detached from the dish (usually 3-4 days).
- Release semiadherent cells from the dish by gentle pipetting. Transfer 3.5 ml of the cell suspension to a sterile vial containing 0.5 ml sterile glycerol. Store at -70°C until you wish to amplify the vector.
- Transfer the remaining 1.5 ml to a microfuge tube and spin 2min at 7000rpm. Aspirate all but about 0.1 ml supernatant. Vortex well to suspend infected cells. To extract DNA, add 0.5 ml pronase working solution to the cells in the microfuge tube and incubate at 37°C for 4-18 h.
- Add 1 ml cold 96% ethanol to precipitate the DNA. Mix well by inverting the tube several times-a fibrous precipitate should be easily visible and the solution should no longer be viscous. Spin 5min at 14,000rpm and then aspirate supernatant. Wash pellet twice with 70% ethanol and air dry.
- Dissolve DNA pellet in 50µl 0.1X SSC by heating at 65°C with occasional vortexing. Digest 5µl with HindIII (1 unit overnight is usually sufficient for complete digestion).
- Apply digested samples and appropriate markers (a HindIII digest of wild-type Ad5 being one convenient marker) to a 1% agarose gel containing ethidium bromide and subject to electrophoresis until the dye front has migrated at least 10cm. If the cytopathic effect was complete, viral DNA bands should be easily visible (under ultraviolet light) above a background smear of cellular DNA. Note that in HindIII digests of human DNA there will be a band of cellular repetitive DNA at 1.8 kb.
- Verify candidate recombinants using other diagnostic restriction enzymes. Although generally 100% of viral plaques obtained using AdMax are correct, it is good laboratory practice to carry out one round of plaque purification, as described later, and screening as described in thissection prior to preparation of high-titer stocks.
Solutions
PBS++ and MEMF11-agarose overlay (at 44°C): See Section III.C.
Steps
- Set up 60-mm dishes of 293 ceils to be confluent at time of infection.
- Remove medium from dishes. Add 0.2ml virus (dilution of agar plug suspension in PBS++ if you wish to plaque purify or dilution of stock for titration). We typically assay dilutions ranging from 10-3 to 10-6 for plaque purification or 10-4 to 10-10 for virus titration. Adsorb the virus for 30-60min in an incubator, occasionally rocking the dishes. Add 10ml MEMF11- agarose overlay, cool, and then continue incubation at 37°C.
- Plaques should be visible within 4-5 days and should be counted for titration at 7 days and again at 10 days. For plaque purification, proceed as for isolation of plaques following transfections (Section III.C).
E. Preparation of High-Titer Viral Stocks (Crude Lysates) from Cells in Monolayer
Because most of the virus remains associated with the infected cells until very late in infection, high-titer stocks can be prepared easily by concentrating infected 293 cells as described here.
Solutions
PBS++, PBS++ + 10% glycerol, and complete MEMF11 + 5% HS: See Section III.C.
- Set up 150-mm dishes of 293 cells to be 80-90% confluent at time of infection. We generally use eight or more dishes for each virus.
- To prepare high-titer stocks, remove medium from the 293 cells and infect at a multiplicity of infection (MOI) of 1-10 PFU per cell (1 ml diluted virus per 150-mm dish). For the initial stock preparation, we dilute virus (from the untitered 4-ml sample stored at -70°C after the last round of viral screening) 1:8 with PBS++. To minimize generation and amplification of rearranged forms of the vector, always prepare hightiter stocks from viral screening samples, not from CsCl-banded stocks (Section III.F).
- Adsorb for 30-60 min and then refeed with complete MEMF11 + 5% HS. Incubate at 37°C and examine daily for signs of a cytopathic effect.
- When the cytopathic effect is nearly complete, i.e., most cells rounded but not yet detached, harvest by scraping the cells off the dish, combining the cells plus spent medium, and centrifuging at 800g for 15min. Aspirate the medium and resuspend the cell pellet in 2ml PBS++ + 10% glycerol per 150-mm dish. Freeze (-70°C) and thaw (37°C) the crude virus stock two or three times prior to titration. Store aliquots at -70°C.
F. Preparation of High-Titer Viral Stocks (Purified) from Cells in Suspension
Recombinant Ads can be purified from crude lysates of either monolayer or suspension cultures. Due to the greater ease of handling suspension cultures, however, this source is preferable for the preparation of purified high-titer viral stocks as described here. Similar yields can be obtained from thirty to sixty 150-mm dishes of 293 cell monolayers.
- Complete Joklik's modified MEM + 10% HS: Add 50ml HS to 450ml complete Joklik's modified MEM (described in Section III.B). Store at 4°C.
- 1% sodium citrate: Dissolve 1 g trisodium citrate dihydrate in H2O to a final volume of 100ml.
- Carnoy's fixative: Add 25 ml glacial acetic acid to 75 ml methanol.
- Orcein solution: Add 1 g orcein dye to 25 ml glacial acetic acid plus 25ml H2O. Filter through Whatman No. 1 paper.
- 0.1M Tris-HCl, pH 8.0: Add 1.2g Tris base to 80ml H2O. Adjust pH to 8.0 with HCl. Adjust volume to 100 ml and autoclave.
- 5% Na deoxycholate: Add 5g Na deoxycholate to 100 ml H2O.
- 2 M MgCl2: Add 40.6 g MgCl2·6H2O to 100 ml H2O and sterile filter.
- DNase I solution: Dissolve 100mg bovine pancreatic deoxyribonuclease I (DNase I) in 10ml of 10mM Tris-HCl, pH 7.4, 50mM NaCl, 1 mM dithiothreitol, 0.1 mg/ml bovine serum albumin, 50% glycerol. Store in small aliquots at -20°C.
- CsCl solutions for banding: Transfer the indicated amounts of analytical grade CsCl into small beakers to give the desired final densities:
Solution Density of final solution (g/ml) Weight of CsCl (g) 1.5d 1.5 83.1 1.35 d 1.35 54.3 1.25 d 1.25 37.0
Add 100 ml 10 mM Tris-HCl, pH 8, to each beaker and stir to dissolve. Verify density by weighing 1.0 ml of each solution (e.g., 1.0ml of the 1.35d solution should weigh 1.35 g). Sterile filter and store at room temperature. - Sterile glycerol: Prepare by autoclaving.
- TE/SDS: Add 0.5ml 20% SDS to 100ml 10mM Tris-HCl, 1 mM EDTA, pH 8.
- Grow 293N3S cells in spinner culture to a density of 2-4 × 105 cells/ml in 3 liters of complete Joklik's modified MEM + 10% HS. Centrifuge cell suspension at 750g for 20min, saving half of the conditioned medium. Resuspend the cell pellet in 0.1 vol fresh medium and transfer to a sterile 500-ml bottle containing a sterile stir bar.
- Add virus at an MOI of 10-20PFU/cell and stir gently at 37°C. After 1 h, return culture to the spinner flask and bring to the original volume using 50% conditioned medium and 50% fresh medium. Continue stirring at 37°C.
- Monitor infection daily by inclusion body staining as follows.
- Remove a 5-ml aliquot from the infected spinner culture. Spin for 10min at 750g and resuspend the cell pellet in 0.5 ml of 1% sodium citrate.
- Incubate at room temperature for 10 min, add 0.5 ml Carnoy's fixative, and fix for 10min at room temperature.
- Add 2ml Carnoy's fixative, spin for 10min at 750 g, aspirate, and resuspend the pellet in a few drops of Carnoy's fixative. Add one drop of fixed cells to a slide, let air dry for about 10min, add one drop orcein solution and a coverslip, and examine in the microscope. Inclusion bodies appear as densely staining nuclear structures resulting from the accumulation of large amounts of virus and viral products at late times in infection. A negative control should be included in initial tests.
- When inclusion bodies are visible in 80-90% of the cells (1.5 to 3 days), harvest by centrifugation at 750g for 20min in sterile l-liter bottles. Combine pellets in a small volume of medium and spin again. Resuspend pellet in 15 ml 0.1M Tris-HCl, pH 8.0. Store at -70°C until use.
- Thaw the frozen crude stock and add 1.5 ml 5% Na deoxycholate. Mix well and incubate at room temperature for 30min. This disrupts cells without disrupting virions, resulting in a relatively clear, highly viscous suspension.
- Add 0.15ml 2M MgCl2 and 0.075ml DNase I solution and then mix well. Incubate at 37°C for 60min, mixing every 10min. The viscosity should be reduced greatly.
- Spin at 3000g for 15min at 4°C in a tabletop centrifuge.
- Prepare three CsCl step gradients in SW41 Ti ultraclear tubes: Add 0.5ml of 1.5d CsCl solution to the bottom of each tube, carefully overlay with 3.0ml of the 1.35 d solution, and then overlay with 3.0ml of the 1.25d solution. Mark the level of the interface between the 1.35d and the 1.25 d layers.
- Carefully add 5 ml of supernatant from step 6 to the top off each gradient. If necessary, top off tubes with 0.1M Tris-HCl, pH 8.0.
- Spin at 35,000rpm in a Beckman SW41 Ti rotor at 10°C for 1 h.
- Collect the virus band at the interface between the 1.35d and the 1.25d layers by piercing the side of the tube with an 18-gauge needle attached to a 5-ml syringe. Pool the virus from all three tubes into a Beckman SW50.1 ultraclear tube. If necessary, top off the tube with 1.35d CsCl solution.
- Centrifuge the pooled virus in a Beckman SW50.1 rotor at 35,000rpm, 4°C for 16-20h.
- Collect the virus band in the smallest volume possible, transfer to a Slide-A-Lyzer dialysis cassette, and dialyze at 4°C against three changes of 500 volumes 10 mM Tris-HCl, pH 8.0, for at least 24 h total.
- After dialysis, add sterile glycerol to a final concentration of 10%. Store the purified virus at -70°C in small aliquots.
- Determine titer of virus by plaque assay (Section III.D).
- The concentration of virus particles, based on DNA content, can be determined spectrophotometrically as follows.
- Dilute purified virus 20-fold with TE/SDS. Set up blank by diluting virus storage buffer (10 mM Tris, pH 8.0, supplemented with glycerol to 10%) 20-fold with TE/SDS.
- Incubate for 10min at 56°C. Vortex sample briefly. Measure OD260 with a spectrophotometer.
- Calculate the number of particles per milliliter, based on the extinction coefficient of wild-type Ad as determined by Maizel et al. (1968) as follows:
particles/ml = (OD260)(20)(1.1 × 1012)
Once the desired recombinant Ad virus is obtained, the ability to express the foreign gene must be tested. The most suitable procedure for detecting expression would depend on the particular properties of the foreign protein. If antibodies to the protein are available, then ELISA, Western blotting analysis, or immunoprecipitation of infected cell extracts may be the simplest method to quantitate protein expression. If possible, the biological activity of the recombinant protein should also be tested to ensure that the expressed protein is functional (for additional details, see Graham and Prevec, 1991).
It is important to use caution when handling recombinant Ads. Experimentation with these vectors should be carried out in accordance with relevant regulations. If exposed inadvertantly, individuals without previous immunity to Ad5 may seroconvert not only against Ad5, but also against the foreign gene product expressed. This should be avoided, especially if the development of antibody may confuse diagnosis of a particular disease. Finally, no toxic or oncogenic gene product should be expressed from replicationproficient Ad vectors without an appropriate increase in biocontainment.
V. PITFALLS
- There can be a number of different causes for failure to obtain the proper recombinant virus following cotransfection. First, the transfection efficiency may be low (a suitable control would be transfection of wild-type viral DNA or infectious plasmid DNA such as pFG140). The 293 cells used in transfections must be at low passage, growing slowly, and slightly subconfluent. In addition, the plasmid DNA must be of high quality; we routinely use CsCl-banded DNA in cotransfections. Finally, although infrequently, the desired recombinant might not be obtained because the foreign gene insert is toxic to the cells or virus, in which case it may be necessary to use an alternate adenovirus rescue system (e.g., Hi-IQ AdMax from Microbix).
- For many applications, particularly those involving in vivo studies or infection of human cells, it is important to confirm that preparations of E1 replacement Ad vectors are not contaminated with replication- competent Ad (RCA). Most often RCA are generated by recombination of the vector with E1 sequences in the 293 cell genome. The latter is most problematic when the wild-type Ad has a growth advantage over the desired recombinant virus. Several tests for RCA contamination have been described, including a functional assay for virus growth in noncomplementing cell lines such as HeLa or A549, as well as protocols that detect contaminating wild-type viral DNA sequences, such as Southern blot hybridization analysis and quantitative polymerase chain reaction amplification (Lochmuller et al., 1994). It is advisable to perform at least one of these tests in order to estimate the maximum possible RCA contamination in recombinant virus stocks before use.
- The level of expression in E1 replacement vectors depends mainly on the strength of the promoter immediately upstream of the coding sequence for the foreign gene. In E3 insertion vectors, this is not necessarily true; even some promoterless constructs express relatively high levels of recombinant proteins. In these cases the major late or E3 promoter is presumably driving expression of the foreign gene insert. It is not, at this time, possible to predict which E3 constructs will utilize an inserted promoter; consequently, care must be taken in analyzing E3 insertion recombinants for appropriate expression, in particular, for example, when transcriptionally regulated expression is required.
We thank Urea Sankar, John Rudy, and Derek Cummings for excellent technical assistance. This work was supported by grants from the National Institutes of Health, the Canadian Breast Cancer Research Initiative, the Canadian Institutes of Health Research (CIHR), and the National Cancer Institute of Canada. P.N. was supported by a CIHR Postdoctoral Fellowship.
References
Addison, C. U, Hitt, M., Kunsken, D., and Graham, E L. (1997). Comparison of the human versus murine cytomegalovirus immediate early gene promoters for transgene expression by adenoviral vectors. J. Gen. Virol. 78, 1653-1661
Berkner, K. L. (1988). Development of adenovirus vectors for expression of heterologous genes. Biotechniques 6, 616-629.
Bett, A. J., Haddara, W., Prevec, L., and Graham, E L. (1994). An efficient and flexible system for construction of adenovirus vectors with insertions or deletions in early regions 1 and 3. Proc. Natl. Acad. Sci. USA 91, 8802-8806.
Bramson, J., Hitt, M., Gallichan, W. S., Rosenthal, K. L., Gauldie, J., and Graham, E L. (1996). Construction of a double recombinant adenovirus vector expressing a heterodimeric cytokine: In vitro and in vivo production of biologically active interleukin-12. Hum. Gene Ther. 7, 333-342.
Ginsberg, H. S. (1984). "The Adenoviruses." Plenum, New York.
Graham, E L., and Prevec, L. (1991). Manipulation of adenovirus vectors. In "Methods in Molecular Biology" (E. J. Murray, ed.), Vol. 7, pp. 109-128. Humana Press, Clifton, NJ.
Graham, F. U, Smiley, J., Russell, W. C., and Nairn, R. (1977). Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J. Gen. Virol. 36, 59-72.
Hitt, M. M., Parks, R. J., and Graham, E L. (1999). Structure and genetic organization of adenovirus vectors. In "'The Development of Human Gene Therapy" (T. Friedmann, ed.), pp. 61-86. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
Lochmuller, H., Jani, A., Huard, J., Prescott, S., Simoneau, P. M., Massie, B., Karpati, E, and Acsadi, G. (1994). Emergence of early region 1-containing replication-competent adenovirus in stocks of replication-defective adenovirus recombinants (AE1 + AE3) during multiple passages in 293 cells. Hum. Gene Ther. 5, 1485-1491.
Maizel, J. V., White, D., and Scharff. M. D. (1968). The polypeptides of adenovirus. I. Evidence of multiple protein components in the virion and a comparison of types 2, 7a, and 12. Virology 36,115- 125.
Ng, P., Cummings, D. T., Evelegh, C. M., and Graham, E L. (2000b). Yeast recombinase FLP functions effectively in human cells for construction of adenovirus vectors. Biotechniques 29, 524-526, 528.
Ng, P., and Graham, E L. (2002). Construction of first-generation adenoviral vectors. Methods Mol. Med. 69, 389-414.
Ng, P., Parks, R. J., Cummings, D. T., Evelegh, C. M., and Graham, E L. (2000a). An enhanced system for construction of adenoviral vectors by the two plasmid rescue method. Hum. Gene Ther. 11, 693-699.
Sambrook, J., and Russell, D. (2001). "Molecular Cloning: A Laboratory Manual" 3rd Ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
Support our developers




