Generation of Human and Murine Dendritic Cells
Dendritic cells (DC) are professional antigenpresenting cells (APC) that serve as sentinels for the induction and regulation of immune responses. Because they are potent stimulators for B and T lymphocytes and activate natural killer cells, they link the innate and the acquired immune system (Fernandez et al., 1999). Dendritic cells are migratory leucocytes originating from the bone marrow, specialized for the uptake, processing, and presentation of antigens. In peripheral tissues, they are found in an immature stage. After contact with antigenic material in the context of danger signals, they mature and upregulate major histocompatibility complexes and costimulatory molecules required for effective interaction with T cells. They are considered a powerful tool to manipulate the immune system (Banchereau and Steinman, 1998).
In mice, DC subsets in different tissues can be distinguished (Shortman and Liu, 2002), e.g., Langerhans cells in skin, three subsets of spleen DC characterized by their CD8 and CD4 expression (Vremec et al., 2000; McLellan and K/impgen, 2000), and at least three subsets of lymph node DC (Ruedl et al., 2000; Vremec et al., 1997). In addition, dendritic cells can be generated from precursors in bone marrow or from peripheral blood monocytes (Inaba et al., 1992; Schreurs et al., 1999).
Acid citrate dextrose (ACD-A, Prod. No. 70010035) can be obtained from Fresenius, Bad Homburg, Germany, and Lymphoprep. (Cat. No. 1053980) from Axis-Shield PoC AS, Oslo, Norway. Phosphatebuffered saline (PBS) (Cat. No. BE17-512F); EDTA, 0.2 mg/ml (Cat. No. 17-711E), RPM11640 (Prod. No. BE12- 167F); Hanks' balanced salt solution (HBSS) (Cat. No. BE10-547F); and L-glutamine, 200mM (Cat. No. BE17- 605E), are available from Bio Whittaker, Cambrex Company, Verviers, Belgium, 20% human serum albumin (HSA) from Aventis Behring, Marburg, Germany, glucose 40% (Prod. No. 2357542) from Braun, Melsungen, Germany, and dimethyl sulfoxide (DMSO) (Prod. No. D-2650), as well as bovine serum albumin (5% stock solution, Prod. No. A-4128), are from Sigma-Aldrich Chemie, Taufkirchen, Germany. For cytokine use, GM-CSF (Leucomax 400) from Essex Pharma, Mfinchen, Germany, interleukin (IL)-4 (Cat. No. 9511231), TNF-α (Cat. No. 9511512), IL-1β (Cat. No. 9511180), and IL-6 (Cat. No. 9511260) from Strathmann, Hamburg, Germany, can be used. PGE2 (Minprostin E2 10mg/ml) is produced by Pharmacia, Erlangen, Germany. Refobacin 10mg is provided by Merck, Darmstadt, Germany, penicillin/streptomycin (PenStrep, Cat. No. 15140-122) by Invitrogen Corporation, Karlsruhe, Germany, and Liquemin N 10000 by Hoffmann-La Roche AG, Grenzach-Wyhlen, Germany. Fetal calf serum (FCS, Cat. No. 14-801E) can be obtained from Bio Whittaker. Syringes, 10ml (Prod. No. 4606108) and 20ml (Prod. No. 4606205), can be obtained from Braun. Culture flasks, 80 cm3 (Prod. No. 201045) and 175cm3 (Prod. No. 200573), as well as cryotubes, 1.8 ml (Prod. No. 363401) and 3.6 ml (Prod. No. 366524), are available from Nunc GmbH & Co. KG, Wiesbaden, Germany. Tubes, 15ml (Prod. No. 25315- 15) and 50ml (Cat. No. 430829), are from Corning GmbH, Bodenheim, Germany. Disposable filter units (red rim, Ref. No. 10462200) are from Schleicher & Schuell, Dassel, Germany. Pipettes, 2ml (Cat. No. 4486), 5 ml (Cat. No. 4487), 10ml (Cat. No. 4488), and 25 ml (Cat. No. 4489), are from Corning GmbH. Pipette tips, 10 µl (Cat. No. 790011), are available from Biozym, Hessisch-Oldendorf, Germany; 100 µ (Cat. No. 0030 010.035) and 1000 µl (Cat. No. 0030 010.051) are from Eppendorf. Tissue culture dishes (Prod. No. 35-3003) are from Falcon, Becton-Dickinson, New Jersey, syringe needles, 0.9 × 40mm (Prod. No. 4657519), are from Braun, trypan blue (Cat. No. $11-004) is from PAA Laboratories, Linz, Austria, and antibodies for FCM analysis are from Becton-Dickinson. Antibodies, antibody-conjugated diamagnetic beads, separation columns, and magnets for the isolation of peripheral blood dendritic cells can be obtained from Miltenyi Biotech GmbH, Bergisch-Gladbach, Germany (blood dendritic cell isolation kit, Cat. No. 130-046-801). For further reference, we recommend the frequently updated online protocols of Miltenyi (http://www. miltenyibiotech.com).
B. Murine Dendritic Cells
Bovine serum albumin (BSA) (Cat. No. B 4287), β-mercapthoethanol (Cat. No. M 6250), and EDTA (Cat. No. 6511) are from Sigma-Aldrich, Deisenhofen, Germany. Cytokines are from Strathmann Biotech, Hamburg, Germany: rmGM-CSF (Cat. No. mGMCSF- 10) and rmIL-4 (Cat. No. mIL4-10). Collagenase type II (Cat. No. LS04176) or type III (Cat. No. LS04182) is from Cell Systems Biotech GmbH, St. Katharinen, Germany. DNase I is from Roche Diagnostics GmbH, Mannheim, Germany (20mg/ml, Cat. No. 92566700). Fetal calf serum (FCS) (Cat. No. 14-801E) was tested before usage and obtained as IMDM (Cat. No. 12-726F) from Cambrex/Bio Witthaker Europe, Verviers, Belgium. Hank's (Cat. No. 14175-053), HBSS (Cat. No. 14175-053), L-glutamine (Cat. No. 25030-024), penicillin/ streptomycin (Cat. No. 15140-122), PBS (Cat. No. 14190094), and RPMI 1640 (Cat. No. 31870-025) are from Invitrogen, Karlsruhe, Germany. Heparin (Cat. No. L6510) and HEPES are from Biochrom AG Seromed, Berlin, Germany (Cat. No. L 1603). KCl (Cat. No. 104933) and NaCl (Cat. No. 106404) are from Merck, Darmstadt, Germany. Trypsin (Cat. No. 59227) is from JRH Biosciences, Shawnee Mission, Kansas. Nycodenz powder (Nycoprep) is from Nycomed Pharma AS, Asker, Norway. Plastic materials are obtained from Becton-Dickinson, Heidelberg, Germany: cell culture dish (Cat. No. 353003), petri dish (Cat. No. 351001), 70-µm cell stainer (Cat. No. 352350), 6-well plate (Cat. No. 353224), 24-well plate (Cat. No. 353226), syringe needle (Cat. No. 300400), and 50-ml tube (Cat. No. 352070). Instruments are from NeoLab, Heidelberg, Germany: sieve (diameter 70mm, Cat. No. 2-7530) and tweezers (Cat. No. 2- 1034). Cy5-PE-conjugated anti-CD4 monoclonal antibody (mab) (clone H129.19; Cat. No. 553654) is from Becton-Dickinson. Multiple species Ig-absorbed, fluorescein isothiocyanate (FITC)-conjugated antihamster Ig (Cat. No. 127-096-160) and PE antirat Ig (Cat. No. 112-116-143) are both from Dianova, Hamburg, Germany.
Solutions for the Generation of Human Dendritic Cells
- PBS/O.1M EDTA buffer: 2.5ml 2M EDTA and 497.5ml PBS. Store at 4°C and use within 1 week.
- Freezing medium: 20% HSA, 20% DMSO, and 6% glucose. To prepare 200-ml, pipette 130ml HSA 20% into a sterile 200-ml flask and add 40ml DMSO and 30ml 40% glucose. Store at 4°C and use within 1 week.
- RO culture medium (RO): To one bottle of 500ml RPMI 1640, add 5ml glutamine and 2ml 10mg Refobacin. Store at 4°C and use within 1 week.
- Complete medium (CM): To one bottle of 500ml RPMI 1640, add 5 ml glutamine, 2 ml 10 mg Refobacin, and 5ml sterile-filtered autologous heat-inactivated plasma. Store at 4°C and use within 1 week.
- PBS/ACD buffer: Add 50ml ACD-A to one bottle (500ml) PBS and use within 1 day.
- PBS/heparin solution: Add 500µl Liquemin N 10000 (5000 IE sodium heparin) to 500ml PBS buffer. Store at 4°C.
- R1O culture medium (RIO): 1% glutamine, 1% PenStrep, and 10% FCS in RPMI 1640. To one bottle of 500ml RPMI 1640, add 5 ml glutamine, 5 ml PenStrep, and 50ml heat-inactivated FCS. Store at 4°C.
- Adherence medium (AM): To one bottle of 500ml RPMI 1640, add 5ml PenStrep, 5ml glutamine, 15ml sterile-filtered plasma surrogate, and 20,000 U GMCSE Store at 4°C and use within 1 day.
- BSA/EDTA buffer (BEB): 0.5% bovine serum albumin and 2mM EDTA. For preparation of 100ml buffer, add 10ml of 5% bovine serum albumin stock solution and 3.2ml 2% EDTA to 86.8 ml PBS. Note: it is necessary to degas buffer prior to use. Store and use at 4°C.
A. Generation of Mature Dendritic Cells from Precursor Cells Obtained from Leukapheresis Products and Whole Blood
The development of techniques to generate large numbers of homogeneous DC populations in vitro from human progenitors allowed their use for research purposes as well as for clinical applications. Protocols established by several different groups describe the generation of DC either from rare, proliferating CD34+ cells or from more frequent, but nonproliferating CD 14+ monocytic cells, which can be both isolated from peripheral blood. Currently, monocyte-derived DC are used more frequently, as no special pretreatment (e.g., systemic application of G-CSF) of the donor is required; moreover, DC yielded from this progenitor population still seem to be more homogeneous and easier to generate with reproducible characteristics; hence we will restrict the provided protocol to this population.
Day 0
- Fill leukapheresis product (200-250ml) into a 1000-ml culture flask and add warm (room temperature) PBS-ACD buffer to a final volume of 480ml.
- Fill 15 ml Lymphoprep. into each of sixteen 50-ml tubes.
- Overlay Lymphoprep. carefully with 30 ml of the cell suspension.
- Spin in a warm centrifuge for 30min (22°C, 300g). Make sure that the centrifuge runs off without brake.
- While the cells are spinning, fill autologous plasma in 50-ml tubes and incubate for 30min in a 56°C hot water bath. Thereafter, spin tubes for 10min (22°C 600g), aliquot supernatants into 15-ml tubes, and freeze at-20°C.
- Take leukapheresis product-containing tubes carefully out of the centrifuge and harvest the interphase into 50-ml tubes containing 15ml of cold PBS/EDTA. Add cold PBS/EDTA buffer to a final volume of 45 ml.
- Spin tubes for 10min (4°C, 200g).
- Remove supernatant, resuspend pellet with cold PBS/EDTA buffer by observing a 2:1 transfer, and spin tubes for 5 min (4°C, 300g).
- Repeat step 8.
- Remove supernatant and resuspend with 40ml cold R0.
- Take an aliquot of all four tubes and dilute each 1:50 for cell counting using a Neubauer chamber. While counting cells, put 50-ml tubes containing cells on wet ice.
- After calculating the amount of cells, devide cells in fractions for immediate replating and for freezing. Spin cells in cold centrifuge for 5 min (4°C, 300g).
- Cells that are not used for immediate generation of DC should be frozen in 20% HSA at 120 × 106 cells per 1.8ml and freezing medium (1:1) in cold 3.6-ml vials using a freezing device on wet ice before they are transferred to a nitrogen-freezing machine to cool down to-150°C. Store frozen cells in liquid nitrogen.
- For immediate generation of DC, spin cells in a cold centrifuge for 5min (4°C, 300g), remove supernatant, and resuspend at 15-25 × 106 cells/ml of CM.
- Preload dishes with 8ml CM. Add 2ml of cell suspension per dish, swing slightly, and transfer dishes into the incubator for at least 30min.
- Subsequently, control adherence. If sufficient (close layer of adherent cells), wash away the nonadherent fraction with warm PBS (room temperature) twice. Add 10ml of warm CM per dish and retransfer into an incubator overnight.
- Take dishes out of the incubator and control cell adherence under a microscope.
- Remove CM from dishes by pipetting carefully.
- Add 9ml of warm CM (room temperature).
- Fill 1 ml CM per dish into a 50-ml tube and add 8000 U GM-CSF and 10,000 U IL-4 per dish.
- Add diluted cytokine mix to dishes, swing slightly, and transfer dishes into an incubator.
Day 5
- Take dishes out of the incubator and control cell adherence under a microscope.
- Carefully collect 5ml of culture supernatant from each dish into a 50-ml tube.
- Add 4ml of room-temperatured CM to each dish.
- Spin 50 ml tubes for 5 min (room temperature 300g).
- Remove supernatant, add 1 ml CM, 8000 U GMCSF, and 10,000 U IL-4 per dish and resuspend.
- Redistribute 1 ml of this suspension to each dish, swing slightly, and transfer dishes into an incubator.
Day 6
- Take dishes out of incubator and control cell morphology under a microscope.
- Remove 1 ml from all dishes by pipetting into a 50-ml tube.
- Add TNF-o~ (10ng/ml cell culture volume), PGE2 (lµg/ml), IL-1β (2ng/ml), and IL-6 (5ng/ml), resuspend, and add equal amounts into each dish. Swing slightly and retransfer dishes to an incubator. Incubate for at least 24h.
- Take dishes out of incubator and control cell morphology under a microscope. Mature DC appear veiled and nonadherent.
- Harvest cells into 50-ml tubes, spin for 5min (room temperature 300g), remove supernatant, and resuspend cells in CM. Remove an aliquot to count cells.
- Determine the phenotype of the cells by FCM analysis (Fig. 1).
2. Protocol to Start from Frozen PBMC Isolated from Leukapheresis Products
Day 0
- Fill 5 ml of cold HBSS in 15-ml tubes.
- Thaw vials with frozen cells in warm water to an extent that a frozen core of cells remains in each tube.
- Dump frozen cells into the 15-ml tubes preloaded with 5 ml HBSS. Use one vial of frozen cells per 15-ml tube.
- Rinse the storage tube once with cold HBSS, pipette suspension into the 15-ml tubes, and fill up with HBSS to a final volume of 13-14 ml.
- Spin in a cold centrifuge for 10min (4°C, 240g).
- Remove supernatants, resuspend pellets with a small amount of cold R0, and pool pellets into one 50-ml tube.
- Spin in a cold centrifuge for 5 min (4°C, 300g).
- Remove supernatant and resuspend pellet with 40ml of cold R0.
- Take an aliquot and count cells.
- Follow steps 14-16 of the protocol described in the previous protocol.
For day 1 to day 7 proceed as described in the previous protocol.
- Transfer buffy coat or whole blood into a culture flask and dilute 1:3 with PBS/heparin solution.
- Fill 15 ml Lymphoprep. into 50-ml tubes.
- Overlay Lymphoprep. carefully with 30ml of the cell suspension.
- Spin in a warm centrifuge for 30 min (22°C, 300g). Make sure that the centrifuge runs off without brake.
- Collect supernatant, further called plasma surrogate, into a 50-ml tube and incubate for 30min in a 56°C hot water bath. Thereafter, centrifuge for 10 min (room temperature 600 g). Save supernatant in a 50-ml tube for preparing AM.
- Harvest interphase carefully to a tube containing 15ml cold PBS/EDTA buffer. Fill up to a final volume of 40 ml using PBS/EDTA buffer.
- Centrifuge for 10 min (4°C, 200g).
- Remove supernatant and resuspend pellet in a small volume of PBS/EDTA buffer. Pool pellets (2:1 transfer) and add cold PBS/EDTA buffer to a final volume of 40ml per tube.
- Centrifuge for 5 min (4°C, 300g).
- Repeat steps 8 and 9.
- Remove supernatant, resuspend pellets in a small volume of AM, and pool cells into one 50-ml tube.
- Remove an aliquot and count cells.
- Load dishes with AM and cell suspension by observing 30-50 × 106 cells per dish. Swing slightly and transfer dishes into the incubator for 60 min.
- If cells adhere sufficiently, gently wash away nonadherent cells with warm PBS (room temperature). Repeat once. Add 10ml warm R10 supplemented with 10,000 U IL-4 and 8000 U GM-CSF per dish and put into an incubator overnight.
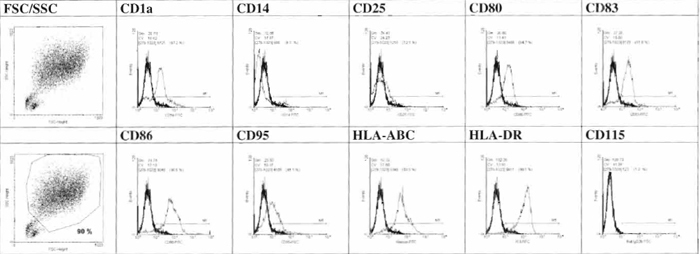 |
| FIGURE 1 Phenotypic characterization of day 7 cell generated by FCM analysis. |
Day 3
- Take dishes out of the incubator and control cell adherence under a microscope.
- Remove 5 ml R10 from dishes by pipetting carefully.
- Add 4ml of warm R10 (room temperature) to each dish.
- Add 4000 U GM-CSF and 5000 IL-4 diluted in 1 ml of R10 to each dish.
- Swing slightly and transfer dishes into the incubator.
Day 5
- Follow steps 1-5 as described for day 3.
Day 7
- Take dishes out of incubator and control cell morphology under a microscope.
- Harvest cells into 50-ml tubes, spin for 5min (4°C, 300g), and collect supernatant. Resuspend cells in R10. Remove an aliquot to count cells.
- Replate 5-7 × 106 cells into each culture dish containing 10ml final volume (1:1 saved supernatant and R10). Add 4000 U GM-CSF and 5000 IL-4 per dish, as well as TNF-α (10ng/ml cell culture volume), PGE2 (lµg/ml), IL-1β (2ng/ml), and IL-6 (5ng/ml), and pipette equal portions into the dishes.
- Swing dishes slightly and transfer them into the ncubator.
- Take dishes out of the incubator and control cell morphology under a microscope. Mature dendritic cells look veiled and nonadherent.
- Harvest cells into 50-ml tubes, spin for 5 min (4°C, 300g), remove supernatant, and resuspend cells in R10.
- Determine the phenotype of cells by FCM analysis.
B. Direct Isolation of Myeloid and Plasmacytoid DC from Peripheral Blood
In principle, peripheral blood DC are obtained by isolation of lineage marker negative, CD4+ cells (O'Doherty et al., 1994). This basic procedure is described in the following text. However, advanced procedures were developed for the isolation of DC subtypes based on blood DC antigen (BDCA) expression; therefore, myeloid DC can be isolated by depletion of B cells from PBMC and positive selection of BDCA-I+ cells; plasmacytoid cells are isolated directly by the selection of BDCA-4 expressing cells. Techniques for the isolation of peripheral blood DC are based on positive and/or negative selection of primary and secondary antibody-conjugated magnetic beadbinding subpopulations in PBMC.
- Isolate PBMC from heparinized whole blood by standard gradient centrifugation with Lymphoprep; about 2 × 108 cells are usually needed. Note: Due to large cell need, i.e., about 1% cell yield at the end of the procedure, we recommend buffy coats.
- Resuspend cells in 300µl of buffer per 1 × 108 cells. Add 100µl human IgG (FcR blocking reagent) and label cells with 100µl haptenized murine anti- CD3, anti-CDllb, and anti-CD16 antibodies (Hapten- Antibody cocktail) per 108 cells. Incubate at 4°C for 10min.
- Wash cells twice with 20-fold labeling volume with buffer and centrifuge at 300g for 10min. Finally, resuspend the pellet in 900µl buffer / 108 cells.
- Add 100µl cells antihapten antibodies attached to diamagnetic beads (antihapten microbeads), per 108 mix well, and incubate for 15 min at 4°C.
- Separate magnetic (T cells, monocytes, and natural killer cells) cells from the nonmagnetic fraction using a magnetic separation column. To do this, a depletion column must be placed into a magnet (we usually use VarioMACS) and must be assembled with a flow resistor (an injection needle of 20-22 gauge is appropriate; follow the manufacturer's instructions) together with a side syringe and a three-way stopcock. Fill the column with degassed cool buffer and rinse. Cool the magnet until use; the procedure itself can be performed at room temperature.
- Apply the cell suspension on top of the depletion column and allowed it to enter for 5min. Wash the column six times and collect the flow through, which contains the desired cells.
- Wash cells at 300g for 10 min. Remove the supernatant and resuspend the cell pellet in 100µl buffer.
- To separate B cells from the DC, which are enriched in the resulting cell fraction, label cells with 100µl anti-CD4-conjugated magnetic beads (MACS CD4 MicroBeads) and incubate for 30min at 4°C.
- Wash cells once by adding 4ml of buffer and centrifuge at 300g for 10min. Resuspend in 500µl of buffer.
- For the next depletion step, a column must be prepared, again following the manufacturer's instruction (we use a MS+ column and a miniMACS magnet). Prepare the column by washing with 500µl buffer.
- Apply and allow cells to penetrate the column.
- Rinse the column with 3 × 500µl buffer (flow through contains nondendritic cells).
- Remove the column from the magnet, place it on a 15-ml plastic tube, and elute cells by rinsing with 1 ml buffer. Use an appropriate plunger to press fluid through the column. Fill the eluate into a fresh column and repeat the procedure.
- Assess DC purity by flow cytometry, with DC defined as HLA-DR+ cells lacking expression of CD3, CD4, CD14, CD16, and CD19.
C. Preparation of Murine Langerhans Cells
This method was published by Kfimpgen et al. (1994) and is described here with some important modifications to improve LC yield and purity.
Solutions
1. Nycodenz gradient (Vremec and Shortman, 1997)
Solution A (Shortman buffer) (500ml of 308 mOsm (EDTA-SS)): 0.154 M NaCl (4.5 g), 4mM KCl (0.1491 g), 14.8 mM HEPES (2.96 ml of 2.5M stock, pH 7.2), and 5mM EDTA (5ml of 0.5M) and make up to 500ml volumetrically with dH2O (Osm = 308 mOsM)
Solution B (230.78ml of 30.55% Nycodenz (d = 1.16)): 70.5 g Nycodenz powder; make up to 230.78 ml volumetrically with dH2O (Osm = 308 mOsM)
2. Langerhans cell culture medium (I10): IMDM, 10% FCS, 50 µM β-mercapthoethanol, 200µM L-glutamine, 100 µg/ml penicillin, and 50 gg/ml streptomycin
Steps
- Kill 15-30 mice by CO2 inhalation, cut off both ears right above the ring cartilage, and place them in a petri dish (Falcon 1001) filled with 10 ml 70% alcohol for 3 rain. Hold ears under alcohol and strike out blood and air with a round side of a bent tweezers. Place ears on a sterile 10 × 10-cm swab and dry them for 20 min at room temperature under a hood.
- Prepare one petri dish containing 6ml HDSS for the inner ear (ventral side with cartilage) and a dish with 9ml HBSS for the soft outer dorsal half of the ear. Split ears by using two Edson forceps and place the corresponding side on top of the HBSS and add 9ml 2.5% trypsin into the dish with the ventral side of the ears and 3.5 ml 2.5% trypsin for the outer halves of the ears. Incubate ventral ear halves for 90min and the dorsal ear halves for 45 min at 37 °C in 5% CO2.
- Place a metal sieve in a petri dish containing 15 ml of cold I10. Peel off the epidermis using two bent curved watchmaker's forceps and lay the epidermal sheets onto the medium. The sheets will tend to spread out upon reaching the surface of the medium. Knock against the sieve for 3 min. Wash cells once prior to an incubation at a density of 1-2 × 107 per 10-ml cell culture dish or 1-2 × 106/ml in a 6-well plate in I10 and 10ng/ml rmGM-CSF at 37°C, 5% CO2 in humidified air for 2-3 days.
- After 2-3 days, cells are harvested and resuspended in 10 ml 14.1% Nycodenz gradient (p = 1.077) before transfer to two 15-ml tubes. This suspension is overlayed with 2ml of Shortman buffer and spun at 4°C at 600g for 20min without the brake. Harvest low-density cells and wash once with medium prior to use.
D. Preperation of Murine Spleen Dendritic Cells
This procedure is performed according to McLellan et al., (2002) with minor modifications.
Solutions
- Nycodenz gradient: See Section A
- Wiirzburger wash buffer: PBS, 1% BSA, 5 mM EDTA, and 20µg/ml DNase
- FACS buffer: PBS and 0.2% BSA
- Dendritic cell culture medium (R10): RPMI 1640, 10% FCS, 50 µM β-mercapthoethanol, 200 µM Lglutamine, 100µg/ml penicillin, and 50µg/ml streptomycin
- Kill five to six mice by CO2 inhalation, disinfect skin with 70% alcohol for I min, cut abdominal skin using sterile siccors below left ribs, and tear away the skin. Cut the peritoneum directly above the spleen, which is isolated by removal of all other tissues. Under laminar air flow, place spleens into 5ml Hanks buffered salt solution in a petri dish. Add 120µg/ml DNase I, pierce splenic capsule at both ends, and with curved watchmaker's forceps squeeze out all cellular contents. Tear the splenic capsule into small pieces, add 400µl FCS to the dish, and transfer the cell suspension and fragments to a 50-ml polypropylene tube and add collagenase to a concentration of 1 mg/ml. Wash plate once with 5 ml Hanks'. Store petri dish after adding another 5-10ml of Hanks' on ice. In a 50-ml tube, pipette cells up and down ten times with a 10-ml pipette to break up loose aggregates. Subsequently, incubate the tube with constant but gentle swirling for 25 min at 37°C.
- After this incubation, add 200 µl 0.5 M EDTA (pH 7.2) and incubate for an additional 5 min. Gently press remaining fragments through a coarse metal sieve with a rubber syringe plunger and collect into the original petri dish.
- Rinse sieve and petri dish with Hanks' and add this to the cell suspension. Filter the cell suspension through a 70-µm cell stainer into a 50-ml tube. Top up to 50ml with cold washing buffer. Wash once.
- Keep tube in ice and gently add to the cell pellet 10ml 14.1% Nycodenz (308mOsm; p = 1.077) for six spleens and slowly resuspend until all clumps are resolved. Transfer the suspension to two 15-ml polypropylene tubes, overlay with 2ml 308mOsm Shortman buffer, and spin at 4°C at 600g for 20min without any brake.
- Harvest low-density cells from all layers without touching the cell pellet and wash once in ice-cold washing buffer. A normal yield should be around 5-10 × 106 CD11c+ DC / spleen at 30-50% purity.
-
- Isolation of spleen DC subsets (CD11c+/CD4- /CD8α-, CD11c+/CD4-/CD8α-, CD11c+/CD4-/CD8α+) by FACS. Block nonspecific Ig-binding sites on lowdensity spleen cells by incubation for 10min in 25µl 10% goat and mouse serum before labeling with 500 µl N418 culture supernatant (hamster antimouse CD11c mab) and 500µl 53-6.7 culture supernatant (rat antimouse CD8α mab) for 30min and ice. After one wash with Wfirzburger buffer, incubate spleen cells with multiple species Ig-absorbed, FITC-conjugated antihamster Ig and PE antirat Ig for 30min on ice in the dark. After an additional washing step, add 400µl 10% rat serum and Cy5-PE-conjugated anti-CD4 mab (clone H129.19); incubate for 30min, subsequently wash cells once, and resuspend at 5 × 107 cells/ml in PBS/1 mM EDTA. Stained cells can now be sorted into CD11c+/CD4-/CD8α-, CD11c+/CD4+/CD8α-, and CD11c+/CD4-/CD8α+ subsets by FACS (e.g., FACS Vantage, Becton-Dickinson, Heidelberg, Germany).
- Enrichment of spleen DC by adherence. Incubate up to 50 × 106 Nycodenz-enriched, low-density cells per each Falcon 3003 dish in R10 supplemented with 10 ng/ml mGM-CSF for 2 h. For 3003 plates, wash gently against the wall of the dish with warm (37°C) R10 about five times. For 24-well plates, wash gently (5 ×) by removal of media with a R10 Pasteur pipette under vacuum and immediately refill each well with R10. Washes must be performed with extreme care to avoid dislodging too many DC. For the final wash, refill wells with 0.5ml R10 with 10ng/ml mGM-CSF (for 24-well plate). The next day, harvest nonadherent DC prior to use in experiments.
E. Preperation of Murine Bone Marrow- Derived Dendritic Cells (BMDC)
This procedure is performed according to Inaba et al. (1992) with minor modifications.
Solution
BMDC culture medium: IMDM, 10% FCS, 50 µM β-mercapthoethanol, 200µM L-glutamine, 100µg/ml penicillin, and 50 µg/ml streptomycin
Steps
- Kill needed number of mice by cervical dislocation or CO2 inhalation, disinfect skin of the legs and lower abdomen with 70% alcohol for I min, cut the skin with a scissors at the inside of the leg upward, tear the skin away, and remove the muscle tissue. Disconnect the complete leg by cutting the ligaments holding the femur in the joint. Scrape the residual tissue with a scalpel from the bone, break it at the distal diaphysis, and transfer the femur and tibia to a tube with PBS.
- Place bones in 70% alcohol for 1 min, wash them with PBS, and place them in a 2-cm petri dish. Cut off both ends of the bones with the scalpel with gently sawing movements; while placing the bone over a 50- ml tube with tweezers, flush the bone marrow with 1 ml of medium using a 10-ml syringe armed with a 0.5 × 24-mm cannula, turn the bone upside down, and repeat flushing.
- Resuspend flushed bone marrow vigorously with a 10-ml pipette for 1 min to dislodge clusters. Transfer the cell suspension into a new 50-ml tube through a 70-µm filter to remove pieces of bone, pellet at 1500rpm for 4min, and resuspend cells. Incubate cells in one 6-well plate/mouse in 3ml medium/well for overnight adherence.
- The following day, harvest and count nonadherent cells. Up to 50% are lost upon adherence. Seed 0.8-1 × 106 cells/well/6-well plate in 4ml BMDC culture medium supplemented with 15ng/ml rmGMCSF and 15 ng/ml rmIL-4.
- On day 3 of culture, add 1 ml medium containing 50ng/ml rmGM-CSF and 50ng/ml rmIL-4 to each well.
- On day 6, after resuspending cells three times with a 5-ml pipette to dislodge DC clusters (adherent and nonadherent), harvest, pellet at 1500rpm for 5 min, and replate cells in 6-well plates with new BMDC culture medium-supplemented cytokines as described earlier.
- On day 8, swirl the plates gently, harvest nonadherent cells (Fig. 2) by gently washing the bottom of the well once, and count cells. The expected yield of BMDC is 10 × 106 cells per mouse, depending on the age of the animal.
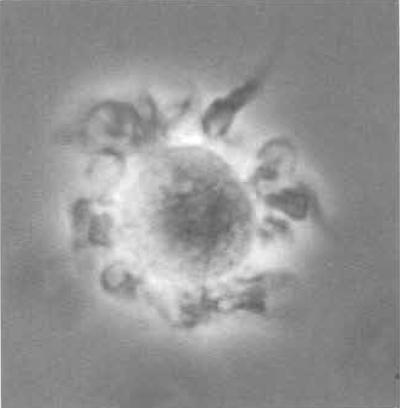 |
| FIGURE 2 Murine bone marrow-derived dendritic cell in culture (400×) showing veils and dendrites spread homogeneously over the cell surface. |
F. Preperation of Murine Monocyte-Derived Dendritic Cells (MoDC)
This procedure is performed according to Schreurs et al. (1999) with little modification.
- Transport medium: ice-cold HBSS (without Ca, Mg) supplemented with 100 U/ml heparin
- Stock solution for mouse monocyte gradient: Nine parts Percoll and one part 10x PBS. Do not use longer than 2 days.
- Working solution for mouse monocyte gradient: Take 18.3ml of the gradient stock solution and add 11.7 ml HBSS; mix well and store at 12 °C.
- MoDC culture medium: IMDM, 10% FCS, 50µM β- mercapthoethanol, 200 µM L-glutamine, 100 µg/ml penicillin, and 50µg/ml streptomycin
- MoDC adherence medium: IMDM, 3% FCS, 50 µM β- mercapthoethanol, 200 µM r-glutamine, 100 µg/ml penicillin, and 50µg/ml streptomycin
- Anesthetize mice one by one, disinfect skin of the upper abdomen with 70% alcohol for 1-min, puncture heart with a 1-ml syringe harnessed with a 0.5 × 24-mm cannula containing 50U heparin, and aspirate blood slowly. Expect 0.7-1ml blood per mouse. Transfer blood to the tube containing transport medium. Immediately afterward kill the mouse by cervical dislocation. In general, 30 mice are needed to obtain sufficient numbers of MoDC precursors. In the following steps, all measures are described for this number of mice.
- Dilute blood to 100 ml in HBSS and overlay carefully in four fractions on a 7-ml mouse monocyte gradient in four 50-ml tubes. Spin for 30min at 2850rpm, 12°C, without any brake. Pool two interphases, wash twice with ice-cold HBSS, pool the remaining pellets, and wash five times with ice-cold HBSS to remove all thrombocytes. Finally wash once with MoDC adherence medium and count cells.
- Resuspend cells in 12ml MoDC adherence medium and incubate for 90min at 37°C in a 6-well plate at 2ml per well for adherence. Discharge nonadherent cells, wash each well four times, and subsequently check the purity of the adherent cells using a microscope at 100× magnification. If satisfactory, add 3ml DC medium/well supplemented with 20ng/ml rmGM-CSF and 20ng/ml rmIL-4.
- After 3 days of culture, add 1 ml medium containing 50ng/ml rmGM-CSF and 50ng/ml rmIL-4 to each well.
- On days 7-9 of culture, swirl the plates gently and harvest nonadherent cells. Gentle washing of the well, which should avoid the detachment of macrophages, improves the yield of cells. Expect 1.5× 106 MoDC per 30 mice.
References
Banchereau, J., and Steinman, R. M. (1998). Dendritic cells and the control of immunity. Nature 392, 245-252.
Celia, M., Facchetti, E, Lanzavecchia, A., and Colonna, M. (2000). Plasmacytoid dendritic cells activated by influenza virus and CD40L drive a potent TH1 polarization. Nature lmmunol. 1, 305-310.
Feuerstein, B., Berger, T. G., Maczek, C., Roder, C., Schreiner, D., Hirsch, U., Haendle, I., Leisgang, W., Glaser, A., Kuss, O., Diepgen, T. L., Schuler, G., and Schuler-Thurner, B. (2000). A method for the production of cryopreserved aliquots of antigenpreloaded, mature dendritic cells ready for clinical use. J. Immunol. Methods 245, 15-29.
Inaba, K., Inaba, M., Romani, N., Aya, H., Deguchi, M., Ikehara, S., Muramatsu, S., and Steinman, R. M. (1992). Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colonystimulating factor. J. Exp. Med. 176, 693-1702.
K/impgen, E., Koch, E, Heufler, C., Eggert, A., Gill, L. L., Gillis, S., Dower, S. K., Romani, N., and Schuler, G. (1994). Understanding the dendritic cell lineage through a study of cytokine receptors. J. Exp. Med. 179, 1767-1776.
McLellan, A. D., and K~impgen, E. (2000). Functions of myeloid and lymphoid dendritic cells. Immunol. Lett. 72, 101-105.
McLellan, A. D., Kapp, M., Eggert, A., Linden, C., Bommhardt, U., Brocker, E. B., Kammerer, U., and Kampgen, E. (2002). Anatomic location and T-cell stimulatory functions of mouse dendritic cell subsets defined by CD4 and CD8 expression. Blood 99, 2084-2093.
Ruedl, C., Koebel, P., Bachmann, M., Hess, M., and Karjalainen, K. (2000). Anatomical origin of dendritic cells determines their life span in peripheral lymph nodes. J. Immunol. 165, 4910-4916.
Schreurs, M. W., Eggert, A. A., de Boer, A. J., Figdor, C. G., and Adema, G. J. (1999). Generation and functional characterization of mouse monocyte-derived dendritic cells. Eur. J. Immunol. 29, 2835-2841.
Shortman, K., and Liu, Y. J. (2002). Mouse and human dendritic cell subtypes. Nature Rev. Immunol. 2, 151-161.
Steinman, R. M., and Pope, M. (2002). Exploiting dendritic cells to improve vaccine efficacy. J. Clin. Invest. 109, 1519-1526.
Thurner, B., Roder, C., Dieckmann, D., Heuer, M., Kruse, M., Glaser, A., Keikavoussi, P., Kaempgen, E., Bender, A., and Schuler, G. (1999). Generation of large numbers of fully mature and stable dendritic cells from leukapheresis products for clinical application. J. Immunol. Methods 1, 1-15.
Vremec, D., Pooley, J., Hochrein, H., Wu, L., and Shortman, K. (2000). CD4 and CD8 expression by dendritic cell subtypes in mouse thymus and spleen. J. Immunol. 164, 2978-2986.
Vremec, D., and Shortman, K. (1997). Dendritic cell subtypes in mouse lymphoid organs: Cross-correlation of surface markers, changes with incubation, and differences among thymus, spleen, and lymph nodes. J. Immunol. 159, 565-573.
Support our developers




