Human Skeletal Myocytes
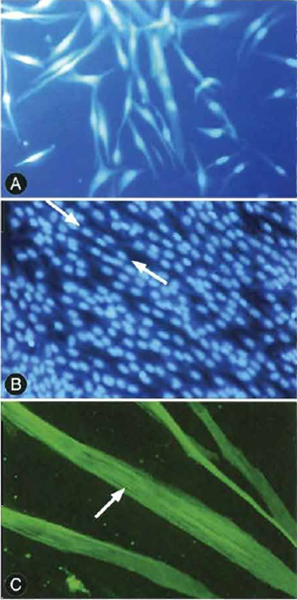 |
| FIGURE 1 Myocytes and myotubules in various stages of differentiation. (A) Individual myocytes prior to differentiation. (B) Myotubules showing the classic "peas in a pod" appearance of multiple nuclei within single cells. Note that there are several mononucleated as well as multinucleated cells in the culture. (C) Striations in differentiated myotubules. |
I. INTRODUCTION
Human muscle tissue consists of many cell types, including adipocytes, fibroblasts, nerve cells, and stromal-vascular components. In the past, biochemical studies of human muscle used organ culture or dissociated monolayers of primary cells. By the nature of theses techniques, both methods would result in contamination of the muscle cells with other cell types. In 1981, Blau and Webster developed a method to maximize proliferation and differentiation of muscle satellite cells to produce cultures of pure human muscle cells. Subsequently, serum-free media were developed to optimize the growth of human myocytes without differentiation (Ham et al., 1988). It then became feasible to use a sequential two-media approach to grow and differentiate human myocytes. With these techniques, and more recent modifications, it is now possible to study human muscle myocytes without the confounding complication of contamination by other cellular components of muscle tissue.
There are many reasons why it is valuable to be able to study myocytes in isolation. Some investigators have considered the potential of human myoblasts in gene therapy. This exploits the important characteristic of muscle cells; the progeny of a single cell can be taken full circle from the animal to the culture dish and then back to the animal where they fuse into mature myofibers of the host (Blau et al., 1993). In fact, this quality of myoblasts affords itself as a way to develop gene- and cell-based therapies for many genetic disorders, including Duchenne muscular dystrophy (Gussoni et al., 1997). A number of investigators have been studying the effects of insulin on multiple aspects of metabolism in human muscle cells as it relates to type 2 diabetes mellitus and other insulin-resistant states (Borthwick et al., 1995; Park et al., 1998; Gaster et al., 2002). Maintenance of muscle cells under controlled conditions permits evaluation of the contribution of the components of the type 2 diabetic environment, such as hyperglycemia, hyperinsulinemia, and hyperlipidemia, to the metabolic behaviors of muscle as compared to the intrinsic or genetic properties of muscle. The culture conditions can also be manipulated to reproduce acquired behaviors. As a final example of how human myoblast cultures can be employed, investigators have also looked at the development of factors during the maturation of human myocytes to myotubes (Gunning et al., 1987). Therefore, the ability to investigate human muscle cells in vitro has a great importance in discovering clinically significant in vivo pathophysiology.
 |
Hams F10 media (Cat. No. 9056), custom ATV (Cat. No. 9920), α-MEM (Cat. No. 9142), Fungibact (Cat. No. 9350), penicillin/streptomycin (pen/strep) (Cat. No. 9366), and glutamine (Cat. No. 9317) are from Irvine Scientific (Santa Ana, CA). The SKBM and bullet kit (Cat. No. 3160) are from BioWhittaker (Walkersville, MD). To make SKGM, the components of the bullet kit are added to one bottle of SKBM, along with 10ml Fungibact and 5 ml glutamine. The insulin bullet can be added or omitted depending on the final purpose for which the cells will be utilized (see later).
Fetal bovine serum (FBS) is from Gemini (Calabasas, CA) (Cat. No. 100-106). The Hams F10 media, SKGM, and α-MEM should all be stored at 4°C. Stocks of custom ATV, fetal bovine serum, glutamine, Fungibact, and pen/strep should be stored at-20°C. To make fusion media, add fetal bovine serum to a final concentration of 2%, 10ml of pen/strep, and 5ml of glutamine to 500ml of α-MEM.
III. PROCEDURE
The following techniques for the growth of human skeletal muscle cultures were established through modifications of previously described methods (Blau and Webster, 1981; Sarabia et al., 1990).
All steps are to be performed under sterile conditions.
- Collect muscle tissue in a 50-ml conical tube in 20ml cold (4°C) Hams F10 media: approximately 100-200mg of tissue is needed.
- Aspirate media. Wash tissue three times with chilled (on ice) Hams F10 to remove blood.
- Transfer tissue in a small volume (~5ml) of Hams F10 to a 100-mm culture dish.
- Mince tissue with sterile scalpels. Make pieces as small as possible.
- Transfer tissue and media back to centrifuge tube and aspirate media with a sterile Pasteur pipette. Because the tissue will settle out on its own, the sample does not need to be centrifuged.
- Add 20ml of trypsin/EDTA (custom ATV) and transfer to a small flask (e.g., 50-ml Erlenmeyer flask) containing a stir bar.
- Stir 20-30min at room temperature in a sterile hood. Collect supernatant and place on ice.
- Add 20ml ATV to flask and repeat step 7 two more times. Pool the supernatants together on ice.
- To the pooled supernatants add FBS to a final concentration of 10%.
- Centrifuge cells for 5 min at 1600 rpm (550g) at room temperature.
- Aspirate supernatant and add 20ml of SKGM [SKBM + bullet kit (with or without insulin) + 10ml Fungibact + 5ml glutamine] to cell pellet. Pipette up and down gently to resuspend cells.
- Transfer media with cells into two 100-mm dishes. Mark plates with subject identifier and place in incubator at 37°C 5% CO2.
- Change SKGM media approximately every 3 days. Continue for 2-3 weeks.
- During the next 2-3 weeks, the muscle cells will grow attached to the surface of the culture dish. Eventuallythe cells will form a confluent layer on the bottom of the dish.
- When the cells are 60-70% confluent, aspirate media and rinse the cells once with 5 ml of custom ATV and aspirate.
- Place another 5 ml custom ATV on plates, aspirate after 30s, and then incubate for 5 min at 37°C.
- Check cells under a microscope to see if they are detached (cells will look rounded).
- If cells are detached, add 5 ml SKGM to rinse plate, collect cells, and transfer to a 15-ml centrifuge tube. If they are only partially detached, remove the attached cells by pipetting up and down gently.
- Add 5ml SKGM media to trypsinized cells and centrifuge at room temperature for 5min at 1600 rpm (550 g).
- Aspirate supernatant and resuspend cells in up to 5 ml SKGM media (not more than 6 ml, depending on volume needed) by pipetting up and down gently.
- Count cells in a hemocytometer chamber.
- Plate cells:
- 100-mm dishes: 60,000 cells per plate (approximately 60 µl cell suspension)
- 6-well dishes: 20,000 cells per well (approximately 30µl)
- 12-well dishes: 6000 cells/well (approximately 20µl)
- 24-well dishes: 3000 cells/well (approximately 10 µl per well)
- Cover wells with SKGM: 10 ml per 100-mm dish, 2ml for each well per 6-well plate, and 1 ml for each well per 12-well and 24-well plates.
- Change the SKGM every 2-3 days.
C. Cell Fusion/Differentiation
- When cell cultures are 80-90% confluent, aspirate media and rinse two times with α-MEM/pen-strep/ 5mM glutamine/2% FCS (fusion media).
- Add fusion media to wells: 10ml per 100-mm dish, 2ml/well per 6-well plate, and 1 ml/well per 12- well and 24-well plates.
- Culture at 37°C, 5% CO2.
- Change media every 48 h.
- Fusion/differentiation is complete by 96 h.
The technique just described has been modified primarily to investigate the metabolic characteristics of human muscle cells. The system has been employed to study insulin action on glucose uptake and glycogen synthesis, fatty acid uptake and oxidation, insulin signaling, and regulation of gene expression. Other investigators have employed different techniques for the culture of human muscle cells; however, the focuses of those investigations were not on hormone action or metabolic activities (Rando and Blau, 1994; Webster and Blau, 1981), which is why the types of media employed differ from those described in this article. The major differences from the procedure described here are that growth media include Hams F-10 with 0.5% chick embryo extract and 20% fetal calf or horse serum. Furthermore, fusion media contain Dulbecco's modified Eagle's media with 2% horse serum. It is uncertain how the constituents of these other media would differ in their impact on human skeletal muscle metabolism when compared to the defined SKGM and fusion media described in this article. In the system described here, cells are passed only a single time before terminal differentiation and are not maintained over multiple passages. Our experience is that both the extent of differentiation (percentage multinucleated cells) and the insulin responsiveness for metabolic events are diminished in a passagedependent manner. Other investigators have maintained cultures from a single subject for a greater number of passages (as high as 15) (Halse et al., 1998).
- It is critical to use sterile technique to avoid cell contamination. Bacteria or fungal contamination can lead to altered metabolic activity of the muscle cells.
- Omit insulin from the SKBM if planning to investigate the effect of insulin on muscle cultures. If the insulin bullet is added, the final concentration (-30µM) is high enough to produce a state of insulin resistance.
- To avoid excessive cell damage from the trypsin, do not overincubate with ATV.
- Change media every 48 h to avoid exhaustion of growth factors and glucose. This can alter the metabolic activity of the muscle culture.
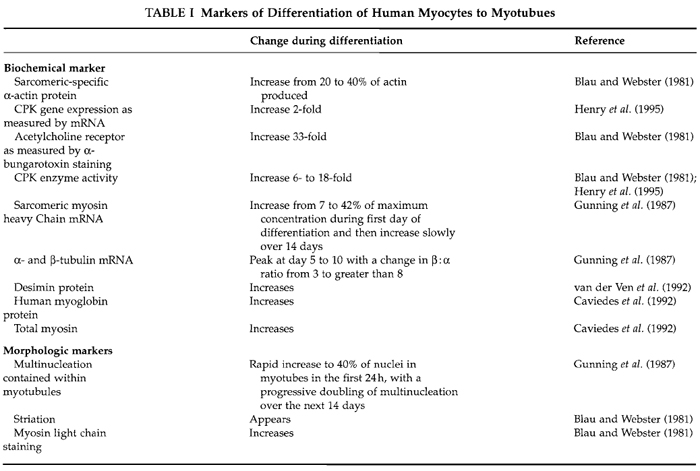
- Treatment of cells can be performed either during the fusion/differentiation period or at completion. If treatment is done during differentiation, then the extent of differentiation must be monitored for each new manipulation. This can be done with careful checking of differentiation by following muscle markers mentioned in Table I.
- It is important to pass the cells at 60-70% confluency. Beyond that point spontaneous fusion may begin in adjacent cells, reducing subsequent proliferation.
- If the fusion media and SKBM are used more than 10 days after assembly, it will be necessary to supplement media with additional glutamine.
Blau, H., Jyotsna, D., and Grace, P. (1993). Myoblasts in pattern formation and gene therapy. TIG 9(8), 269-274.
Blau, H., and Webster, C. (1981). Isolation and characterization of human muscle cells. Proc. Natl. Acad. Sci. USA 78(9), 5623-5627.
Borthwick, A., Wells, A., Rochford, J., Hurel, S., Turnbull, D., and Yeaman, S. (1995). Inhibition of glycogen synthase kinase-3 by insulin in cultured human skeletal muscle myoblasts. Biochem. Biophys. Commun. 210(3), 738-745.
Caviedes, R., Liberona, J., Hidalgo, J., Tascon, S., Salas, K., and Jaimovich, E. (1992). A human skeletal muscle cell line obtained from an adult donor. Biochem. Biophys. Acta 1134(3), 247-255.
Gaster, M., Petersen, I., Hojlund, K., Poulsen, P., and Beck-Nielsen, H. (2002). The diabetic phenotype is conserved in myotubules established from diabetic subjects: Evidence for primary defects in glucose transport and glycogen synthase activity. Diabetes 51(4), 921-927.
Gunning, P., Hardeman, E., Wade, R., Ponte, P., Bains, W., Blau, H., and Kedes, L. (1987). Differential patterns of transcript accumulation during human myogenesis. Mol. Cell. Biol. 7(11), 4100-4114.
Gussoni, E., Blau, H., and Kunkel, L. (1997). The fate of individual myoblasts after transplantation into muscles of DMD patients. Nature Med. 3(9), 970.
Ham, R., St. Clair, J., Webster, C., and Blau, H. (1988). Improved media for normal human muscle satellite cells: Serum-free clonal growth and enhanced growth with low serum. In Vitro Cell. Dev. Biol. 24(8), 833-844.
Henry, R., Abrams, L., Nikoulina, S., and Ciaraldi, T. (1995). Insulin action and glucose metabolism in nondiabetic control and NIDDM subjects. Diabetes 44, 936-946.
Park, K., Ciaraldi, T., Lindgren, K., Abrams-Carter, L., Mudaliar, S., Nikoulina, S., Tufari, S., Veerkamp, J., Vidal-Puig, A., and Henry, R. (1998). Troglitazone effects on gene expression in human skeletal myocytes of type II diabetes involve upregulation of peroxisome proliferator-activated receptor-γ. J. Clin. Endrocrinol. Metab. 83(8), 2830-2835.
Rando, T., and Blau, H. (1994). Primary mouse myoblast purification, characterization and transplantation for cell-mediated gene therapy. J. Cell Biol. 125(6), 1275-1287.
Sarabia, V., Lam, L., Burdett, E., Leiter, L.A., and Klip, A. (1990). Glucose uptake in human and animal muscle cells in culture. Biochem. Cell Biol. 68, 536-542.
Van der Ven, P.E, Schaart, G., Jap, P.H., Sengers, R.C., Stadhounders, A.M., and Ramaekers, EC. (1992). Differentiation of human skeletal muscle cells in culture: Maturation as indicated by titin and desmin striation. Cell Tissue Res. 270(1), 189-198.
Support our developers




