In Situ Electroporation of Radioactive Nucleotides: Assessment of Ras Activity or 32p Labeling of Cellular Proteins
Binding to nucleotide(s) can determine a state of activation of a protein; a number of GTP-binding proteins such as Ras exist in two distinct, guanine nucleotide-bound conformations, the active Ras.GTP state and the inactive Ras.GDP form, so that the fraction of Ras bound to GTP (percentage Ras.GTP/GTP + GDP) can determine its state of activation (Lowy and Willumsen, 1993). Several indirect assays are in existence for measurement of Ras activity (Scheele et al., 1995; Taylor et al., 2001). However, in a number of instances, a direct measurement of Ras.GTP binding is necessary (Egawa et al., 1999) and is commonly performed through the addition of [32P]orthophosphate to the growth medium followed by Ras immunoprecipitation and guanine nucleotide elution (Downward, 1995). This approach is relatively inefficient due to the fact that the isotope is incorporated into all phosphatecontaining cellular components. To circumvent this problem, [α32P]GTP has been introduced into the cell and its breakdown into [α32P]GDP after Ras binding monitored as described earlier (Downward, 1995). Because, contrary to free bases or nucleosides, most nucleotides do not cross the cell membrane, [α32P]GTP has to be introduced into intact cells after cell membrane permeabilization.
This article describes a technique where the introduction of nucleotides into adherent cells is performed through in situ electroporation. Cells are grown on a glass surface coated with electrically conductive, optically transparent indium-tin oxide at the time of pulse delivery, a coating that promotes excellent cell adhesion and growth. Unlike other techniques of cell membrane permeabilization, such as streptolysin- O (SLO) treatment, in situ electroporation does not detectably affect cellular metabolism, presumably because the pores reseal rapidly so that the cellular interior is restored to its original state; under the appropriate conditions there is no increase in the activity of the extracellular signal-regulated kinase (Erk½) or two stress-activated kinases, JNK/SAPK or p38hog (Brownell et al., 1998). Results show that Ras activity measurement through electroporation of [α32P]GTP could be performed using approximately 50-100 times lower amounts of radioactivity; although the 32P is in the form of [α32P]GTP exclusively, this technique offers higher specificity compared to labelling through the addition of [32P]orthophosphate to the culture medium. In addition, labelling of two viral phosphoproteins, the large tumor antigen of simian virus 40 and adenovirus EIA, by in situ electroporation of [γ32P]ATP requires a fraction of the amount of radioactive phosphorus, while offering enhanced specificity.
Dulbecco's modification of Eagle's medium (DMEM) is from ICN (Cat. No. 10-331-22). Phosphatefree DMEM (Cat. No. D-3916) is from Sigma. Fetal calf serum (Cat. No. 2406000AJ) is from Life Technologies Inc. Calf serum is from ICN (Cat. No. 29-131-54). The following reagents are from Sigma: insulin (Cat. No. 1-6643), NaCl (Cat. No. S-7653), HEPES (Cat. No. H-9136), Lucifer yellow CH dilithium salt (Cat. No. L- 0259), trypsin (Cat. No. T-0646), MgCl2 (Cat. No. M- 8266), Triton X-100 (Cat. No. T-6878), deoxycholate (Cat. No. D-5760), EDTA (Cat. No. E-5134), EGTA (Cat. No. E-3889), phenylmethylsulfonyl fluoride (PMSF, P- 7626), aprotinin (Cat. No. A-6279), leupeptin (Cat. No. L-2023), benzamidine (Cat. No. B-6506), dithiothreitol (DTT, Cat. No. D-9779), CaCl2 (Cat. No. C-4901), Trisbase (Cat. No. T-6791), sodium orthovanadate (S-6508), and LiCl (Cat. No. L-4408). The following reagents are from BDH: Extran-300 detergent (Cat. No. $6036 39), SDS (Cat. No. 44244), TLC silica gel plates containing a fluorescence indicator (Cat. No. M05735-01), isopropanol (Cat. No. ACS720), concentrated ammonia solution (Cat. No. ACS033-74), Nonidet P-40 (Cat. No. 56009), and glycerol (Cat. No. B10118). GTP (Cat. No. 106 372) and GDP (Cat. No. 106 208) are from Boehringer Mannheim. The monoclonal anti-Ras antibody is from Oncogene Science (Pan-Ras Ab2, Cat. No. OP22), the monoclonal antibody to simian virus 40 large tumor antigen is from Pharmingen (p108, Cat. No. 14121A), and the monoclonal antibody to adenovirus EIA is from Calbiochem (M73, Cat. No. DPll). Staph. A Sepharose beads (17-0780-03) for immunoprecipitation are from Pharmacia. X-ray film is from Kodak (X-OMAT AR, Cat. No. 165 1454). CelTakTM (Cat. No. 354240) is from BD Biosciences. Tissue culture petri dishes (6cm diameter) are from Corning or Sarstedt.
The apparatus for electroporation in situ (Epizap model EZ-16) is available from Ask Science Products Inc. (487 Victoria St. Kingston, Ontario Canada). The inverted, phase-contrast and fluorescence microscope, equipped with a filter for Lucifer yellow (excitation: 435, emission: 530), is from Olympus (Model IX70).
The technique can be applied to a large variety of adherent cell types. We have used a number of lines, such as the Fisher rat fibroblast Flll and its polyoma or simian virus 40 virus-transformed derivatives, mouse fibroblast NIH 3T3, mouse Balb/c 3T3, mouse NIH 3T6, mouse C3H10T½ fibroblast derivatives expressing a ras-antimessage [e.g., lines R14 and 25B8 (Raptis et al., 1997)], Rasleu61-transformed 10T½, and rat liver epithelial T51B, as well as a variety of differentiated adipocytes (Brownell et al., 1996). All cells can be grown in plastic petri dishes in DMEM supplemented with 5% calf serum in a humidified 5% CO2 incubator with the exception of R14 and 25B8, which are grown in DMEM supplemented with 10% fetal calf serum. Cells that do not adhere can be grown and electroporated on the same conductive slides coated with CelTakTM, poly-L-lysine or collagen.
The apparatus for in situ electroporation is described in Fig. 1. Cells are grown on conductive and transparent glass slides, which are placed in a petri dish to maintain sterility. The cell growth area is defined by a "window" formed with an electrically insulating frame made of Teflon. The pulse is transmitted through a stainless-steel negative electrode, which is slightly larger than the cell growth area and is placed on top of the cells, resting on the Teflon frame. Another stainless-steel block is used as a positive contact bar. A complete circuit is formed by placing the electrode set on top of the slide as shown in Figs. 1A and lB. The frame creates a gap between the conductive coating and the negative electrode so that current can only flow through the electroporation fluid and cells growing in the window. In order to obtain a uniform electric field strength over the entire area below the negative electrode, despite the fact that the conductive coating exhibits a significant amount of electrical resistance, the bottom surface of the negative electrode must be inclined relative to the glass surface, rising in the direction of the positive contact bar, in a manner proportional to the resistance of the coating (Raptis and Firth, 1990); glass with a surface resistivity of 2Ω/sq requires an angle of 1.5°, whereas glass of 20Ω/sq requires an angle of 4.4°. The procedure described is for glass with a surface resistivity of 20Ω/sq, which is readily available and relatively inexpensive, hence it can be discarded after use to limit exposure to radioactivity (see Comment 1). Similarly, the electrode can be made out of inexpensive aluminum for a single use.
![FIGURE 1 Electroporation electrode assembly. (A) Side view. A Delrin carrier (7) holds the negative electrode (3) and the positive contact bar (4) so that they form one unit, which can be placed on top of the slide (1) with its frame (2) and cells (5) in place. Negative (-) and positive (+) signs indicate the electrical connecting points via which the pulse of electricity is delivered to the electrodes from the pulse generator. The underside of the negative electrode (3) is machined to a slight angle, which compensates for the surface resistivity of the conductive slide to provide uniform electroporation of the whole cell growth area. The negative electrode rests on a Teflon frame (2), which insulates it from the conductive surface. The fluid containing the material to be electroporated just fills the cavity below the negative electrode. When a capacitor is discharged, current passes through the electroporation fluid (6) and cells (5) attached to the conductive glass slide (1) to the positive contact bar (4) and back to the pulse source. Note that the angle of the negative electrode has been exaggerated to better illustrate the meniscus of the radioactive electroporation solution (6). The slide and electrode fit in a 6cm petri dish (8) that is locked in place on a stand (9). The top plate supports the petri dish and is made of transparent acrylic so that the operator can look in the mirror (10) to ensure that the liquid (6) is properly filling the cavity without air bubbles. (B) Top view. The outline of the conductive slide (1) with a Teflon frame (2) in place to define the area of cell growth and electroporation are indicated [from Brownell et al. (1997), reprinted with permission]. Upscaling. (C) Side view. Cells to be electroporated (5) are grown on a glass slide (1), coated with ITO (1a). The negative electrode (3a and 3b) is a narrow steel bar mounted across the width of the slide, resting on the Teflon frame (2), which is moved across the surface of the slide as shown by the arrow, by an insulated carrier. The underside of the negative electrode is curved in both directions, such as to optimise the uniformity of electrical field. Note that only the area of cells immediately below the electrode is electroporated by a given pulse. The curvature of the negative electrode has been exaggerated to better illustrate its contour. Note that due to the narrow shape of the electrode, air bubbles do not get trapped easily under it, hence a stand with a mirror may not be necessary. (D) Top view. The outline of the conductive slide with a Teflon frame (2) in place to define the area of cell growth and electroporation and the counterelectrodes (4) are indicated. The assembly is placed in a 10cm petri dish (8) [from Raptis et al. (2003) and Tomai et al. (2003), reprinted with permission].](images/v2_pb_s08_c43_f01.jpg) |
| FIGURE 1 Electroporation electrode assembly. (A) Side view. A Delrin carrier (7) holds the negative electrode (3) and the positive contact bar (4) so that they form one unit, which can be placed on top of the slide (1) with its frame (2) and cells (5) in place. Negative (-) and positive (+) signs indicate the electrical connecting points via which the pulse of electricity is delivered to the electrodes from the pulse generator. The underside of the negative electrode (3) is machined to a slight angle, which compensates for the surface resistivity of the conductive slide to provide uniform electroporation of the whole cell growth area. The negative electrode rests on a Teflon frame (2), which insulates it from the conductive surface. The fluid containing the material to be electroporated just fills the cavity below the negative electrode. When a capacitor is discharged, current passes through the electroporation fluid (6) and cells (5) attached to the conductive glass slide (1) to the positive contact bar (4) and back to the pulse source. Note that the angle of the negative electrode has been exaggerated to better illustrate the meniscus of the radioactive electroporation solution (6). The slide and electrode fit in a 6cm petri dish (8) that is locked in place on a stand (9). The top plate supports the petri dish and is made of transparent acrylic so that the operator can look in the mirror (10) to ensure that the liquid (6) is properly filling the cavity without air bubbles. (B) Top view. The outline of the conductive slide (1) with a Teflon frame (2) in place to define the area of cell growth and electroporation are indicated [from Brownell et al. (1997), reprinted with permission]. Upscaling. (C) Side view. Cells to be electroporated (5) are grown on a glass slide (1), coated with ITO (1a). The negative electrode (3a and 3b) is a narrow steel bar mounted across the width of the slide, resting on the Teflon frame (2), which is moved across the surface of the slide as shown by the arrow, by an insulated carrier. The underside of the negative electrode is curved in both directions, such as to optimise the uniformity of electrical field. Note that only the area of cells immediately below the electrode is electroporated by a given pulse. The curvature of the negative electrode has been exaggerated to better illustrate its contour. Note that due to the narrow shape of the electrode, air bubbles do not get trapped easily under it, hence a stand with a mirror may not be necessary. (D) Top view. The outline of the conductive slide with a Teflon frame (2) in place to define the area of cell growth and electroporation and the counterelectrodes (4) are indicated. The assembly is placed in a 10cm petri dish (8) [from Raptis et al. (2003) and Tomai et al. (2003), reprinted with permission]. |
A large number of signal transducers are present in small amounts in the cell so that a large number of cells may be required to obtain a strong signal. Uniform electroporation of a cell growth area of 32 × 10 mm u s i n g 20 Ω/sq glass (i.e., an angle of 4.4°) and a Teflon frame with a thickness of 0.279 mm using the assembly in Figs. 1A and 1B requires a volume o f ~280 µl. Simple scale-up of this assembly, e.g., to a cell growth area of 50 × 30mm, which can be accommodated in a standard, 10 cm petri dish, is faced with the problem of burning the ITO coating that occurs at the higher voltage and capacitance settings required to electroporate this larger area because of the resistance generated by the greater distance the current has to travel. In addition, the volume required , ~1.7 ml, cannot be held in place by surface tension, while the cost of purchase and disposal of the isotope can be prohibitive for certain experiments. These problems can be solved by using an assembly with a narrow, moveable electrode that electroporates a "strip" of cells at a time (Figs. 1C and 1D); in this configuration, only cells immediately below the negative electrode are electroporated by a given pulse of electricity. After electroporation of the first strip of cells, the electrode is translocated laterally, dragging the solution under it by surface tension so that a new strip of cells is electroporated using mostly the same solution (Figs. 1C and 1D). The electric circuit formed during pulse delivery starts at the negative electrode, passes through the electroporation fluid, the cells and the conductive slide surface, to the two positive contact bars, one on each side of the slide. The two positive contact bars form parallel circuit paths, both carrying current from the conductive surface. To compensate for the resistance of the coating, the bottom surface of the negative electrode must be inclined toward each of the positive contact bars; a 25 mm radius on the bottom of the electrode produces successive strips of even electroporation over the entire cell growth area. Using this assembly, an area of 32 × 10 mm can be electroporated using less than 50 µl of solution with a 2.5 mm-wide electrode, whereas an area of 50 × 30 mm can be electroporated effectively in four, 8 mm-wide strips, with a total of 200 µl of solution.
Solutions
- Lucifer yellow solution, 5mg/ml: To make 1 ml, add 5mg Lucifer yellow to 1 ml phosphate-free DMEM.
- [α32P]GTP: Prepare a solution of 500-2000µCi/ ml in phosphate-free DMEM (see Comment 2).
- Ras extraction buffer: 50mM HEPES, pH 7.4, 150mm NaCl, 5 mM MgCl2, 1% Triton, 0.5% deoxycholate, 0.05% SDS, 1 mM EGTA, 1 mM PMSF, 10 µg/ml aprotinin, 10µg/ml leupeptin, 10mm benzamidine, and 1 mM vanadate. The stock solutions can be made ahead of time. To make 50ml, add 2.5ml of HEPES stock, 1.5ml of NaCl stock, 0.25ml of MgCl2 stock, 0.5 ml Triton, 0.05 g deoxycholate, 0.25 ml of SDS stock, and 0.5ml of EGTA stock and then the protease inhibitors, 0.5ml of PMSF stock solution, 0.05 ml of aprotinin stock, 0.05ml of leupeptin stock, 0.5ml of benzamidine stock, and 0.05 ml of vanadate stock and bring the volume to 50 ml with distilled H2O on the day of the experiment.
10% sodium dodecyl sulfate stock solution: Dissolve 100 g in 1 liter H2O. Store at room temperature. Stable for more than a year.
5 M NaCl stock solution: Dissolve 292.2 g NaCl in 1 liter distilled H2O. Autoclave and store at room temperature. Stable for more than a year.
1M MgCl2 stock solution: Dissolve 203.3 g MgCl2·6H2O in 1 liter H2O. Autoclave and store at room temperature. Stable for more than a year.
100mM EGTA stock solution: Dissolve 38.04 g in 800ml distilled H2O. Adjust pH to 8.0 with NaOH and complete volume to 1 liter with distilled H2O. Stable for more than a year at room temperature.
100mM PMSF stock solution: Dissolve 17.4mg PMSF in 10ml isopropanol and store in aliquots at -20°C. Stable for several months.
10mg/ml aprotinin stock solution: Dissolve 100 mg in 10ml of 0.01M HEPES, pH 8.0. Aliquot and store at -20°C. Stable for several months.
10 mg/ml leupeptin stock solution: Dissolve 100mg in 10ml distilled H2O. Aliquot and store at -20°C. Stable for several months.
1M benzamidine stock solution: Dissolve 1.56 g in 10ml distilled H2O. Aliquot and store at -20°C. Stable for several months.
1M vanadate stock solution: Dissolve 1.84 g in 10ml distilled H2O. Aliquot and store at -20°C. Stable for several months. - Guanine nucleotide elution buffer: 2mM EDTA, 2mM DTT, 0.2% SDS, 0.5 mM GTP, and 0.5 mM GDP.
1M dithiothreitol stock solution: Dissolve 3.09g DTT in 20ml of 0.01 sodium acetate (pH 5.2). Store in 1 ml aliquots at -20°C. Stable for more than a year.
0.5M EDTA, pH 8.0 stock solution: Add 186.1g disodium ethylenediaminetetraacetate 92 H2O to 800 ml distilled H2O. To dissolve, adjust the pH to 8.0 with NaOH pellets while stirring. Bring volume to 1 liter. Stable for more than a year at room temperature.
10 mM GTP stock solution: Dissolve 52.3 mg in 10 ml distilled H2O and store at -20°C. Stable for a month.
10mM GDP stock solution: Dissolve 44.3mg in 10ml distilled H2O and store at -20°C. Stable for a month.
See earlier list for other required stock solutions: To make 500µl of elution buffer, add 10µl of 10% SDS stock solution, 2µl of EDTA stock solution, 1 µl of DTT stock solution, 25µl each of GTP and GDP stock solutions, and bring volume up to 500 µl with H2O. - Thin-layer chromatography (TLC) running buffer: 66% isopropanol and 1% concentrated ammonia. To make 100 ml, mix 66 ml isopropanol, 1 ml concentrated ammonia solution, and 33ml distilled H2O in a fume hood. Make fresh the day of the experiment. Depending on the size of the chromatography tank, this volume may need to be increased.
The appropriate institutional regulations for isotope use must be followed for all experiments.
- Choice of slides. Cell growth areas of 32 × 12 mm are sufficient for most cases. However, for some lines with low Ras levels, a larger number of cells may be required to obtain an adequate signal. In this case, the assembly in Figs. 1C and 1D (cell growth area, 50 x 30mm) may be used. Make sure the glass is clean and free of fingerprints (see Comment 5).
- Plate the cells. Uniform spreading of the cells is very important, as the optimal voltage depends in part on the degree of cell contact with the conductive surface (see Comment 3). Place the sterile glass slide inside a 6cm (for 32 × 10mm) or 10cm (for 50 x 30 mm) petri dish. Add a sufficient amount of medium to cover the slide (approximately 9 ml for a 6 cm dish). Pipette the cell suspension in the window (Fig. 1) and place the petris in a tissue-culture incubator.
- Starve the cells from phosphates by placing them in phosphate-free DMEM and the required amounts of dialysed serum for 2-3h or overnight, depending on the experiment.
- Prior to pulse application, remove the growth medium and wash the cells gently with phosphatefree DMEM.
- Carefully wipe the Teflon frame with a folded Kleenex tissue to create a dry area on which a meniscus can form (see Pitfall 1).
- Add the [α32P]GTP solution. The volume of the solution under the electrode varies with the electroporation assembly and cell growth area. For the setup in Fig. 1A and a cell growth area of 32 × 10mm, the volume is ~280µl, whereas the assembly in Fig. 1C requires ~200µl for a cell growth area of 50 × 30mm. Depending on the exact concentration, this volume will contain ~200µCi [(α32P]GTP in phosphate-free DMEM.
- Carefully place the electrode on top of the cells. Make sure there is a sufficient amount of electroporation buffer under the positive contact bar to ensure electrical contact. Make sure there are no air bubbles between the negative electrode and the cells by looking in the mirror. If necessary, the electrode can be sterilized with 70% ethanol before the pulse, and the procedure carried out in a laminar-flow hood, using sterile solutions.
- Apply three to six pulses of the appropriate strength (40-200V, see Comment 3) from a 10- or 20-µF capacitor, depending on the apparatus used (Fig. 1A vs Fig. 1C).
- Remove the electrode set. Because usually only a small fraction of the material penetrates into the cells, the [α32P]GTP solution can be carefully aspirated and used again.
- Add phosphate-free medium containing dialysed serum if permitted by the experimental protocol and incubate the cells for the desired length of time (see Comment 4).
- Remove the unincorporated material: wash the cells twice with phosphate-free medium lacking serum.
- Extract the proteins. Add 1 ml of extraction buffer to the window area of the slide. Scrape the cells using a rubber policeman into a 15 ml tube and rock the tubes on ice for 20min. Centrifuge for 30min at 1,000rpm in a Beckman J-6 centrifuge to clarify. Preclear the lysates by adding 100µl packed Staph. A-Sepharose beads, incubating on ice for 1 h, and centrifuging for 5 min at 1,000rpm in a Beckman J-6 centrifuge.
- Immunoprecipitate Ras. Incubate the precleared supernatant overnight with pan ras Ab2 antibody bound to Staph. A Sepharose beads while rocking on ice.
- Wash the immunoprecipitate four times with 1 ml of extraction buffer lacking the inhibitors. Use a Hamilton syringe to completely remove all traces of wash solution.
- Elute GTP and GDP off the beads by adding 10-20µl elution buffer to the beads and incubating at 68°C for 20min.
- Spot the eluate containing the labelled nucleotides on a silica gel TLC plate containing a fluorescence indicator. Spot 1 µl each of the stock GTP and GDP solutions to serve as cold standards, easily visible under UV light. Develop the plate using a solution of 1% ammonia-66% isopropanol for about 3-4h.
- Dry the TLC plate, expose to Kodak X-OMAT AR film, and excise the spots for liquid scintillation counting or submit to phosphorimager analysis (see Fig. 2).
![FIGURE 2 Assessment of Ras activity through electroporation of [α32P]GTP. Cells were grown on conductive glass (cell growth area, 32 × 10mm, Figs. 1A and 1B) and starved from serum and phosphates. A solution containing [α32P]GTP was added to the cells and introduced with three pulses delivered from a 20µF capacitor at different voltages as indicated. Cells were subsequently placed in a humidified 37°C, CO2 incubator for 3 h. Ras was extracted and precipitated with the pan-ras Ab2 monoclonal antibody, the bound GTP and GDP eluted and separated by thin-layer chromatography (see text). The plate was exposed for 15h to Kodak XAR-5 film with an intensifying screen. In all panels, arrows point to the positions of cold GTP and GDP standards, respectively. (A) Electroporation does not induce a rapid breakdown of intracellular GTP. Lanes 1-5: Mouse 10T½ fibroblasts were electroporated using voltages of 140-200 V as indicated in the presence of 5µCi [α32P]GTP. Three hours later, nucleotides in a 2µl aliquot of each clarified lysate were separated as described earlier without Ras immunoprecipitation. Lane M: As a marker, an aliquot of the [α32P]GTP used in this experiment was run in parallel. (B) Assessment of Ras activity through electroporation of [α32P]GTP in normal 10T½ and their ras-transformed counterparts. Lanes 1-10:10T½ cells (lanes 1-5) or their rasval12-transformed counterparts, 2H1 (lanes 6-10), were electroporated using voltages of 120-190 V as indicated in the presence of 66 or 33µCi [α32P]GTP, respectively. Proteins were extracted, and the Rasbound GTP and GDP were separated as described earlier. As a control (lane 11), 2H1 cells growing in a 3cm petri to were metabolically labelled with 200 µCi [α32P] orthophosphate and processed as described previously. (C) Assessment of Ras activity in normal 10T½ fibroblasts and their ras-transformed counterparts, 2H1, using the standard SLO permeabilization assay. This assay was performed as described (Brownell et al., 1997) and is shown here as a comparison. Lane 1, 10T½; lane 2, rasval12-transformed, 10T½-derived line 2H1; and lane 3, 2H1 precipitated with control rat IgG instead of anti-Ras antibodies.](images/v2_pb_s08_c43_f02.jpg) |
| FIGURE 2 Assessment of Ras activity through electroporation of [α32P]GTP. Cells were grown on conductive glass (cell growth area, 32 × 10mm, Figs. 1A and 1B) and starved from serum and phosphates. A solution containing [α32P]GTP was added to the cells and introduced with three pulses delivered from a 20µF capacitor at different voltages as indicated. Cells were subsequently placed in a humidified 37°C, CO2 incubator for 3 h. Ras was extracted and precipitated with the pan-ras Ab2 monoclonal antibody, the bound GTP and GDP eluted and separated by thin-layer chromatography (see text). The plate was exposed for 15h to Kodak XAR-5 film with an intensifying screen. In all panels, arrows point to the positions of cold GTP and GDP standards, respectively. (A) Electroporation does not induce a rapid breakdown of intracellular GTP. Lanes 1-5: Mouse 10T½ fibroblasts were electroporated using voltages of 140-200 V as indicated in the presence of 5µCi [α32P]GTP. Three hours later, nucleotides in a 2µl aliquot of each clarified lysate were separated as described earlier without Ras immunoprecipitation. Lane M: As a marker, an aliquot of the [α32P]GTP used in this experiment was run in parallel. (B) Assessment of Ras activity through electroporation of [α32P]GTP in normal 10T½ and their ras-transformed counterparts. Lanes 1-10:10T½ cells (lanes 1-5) or their rasval12-transformed counterparts, 2H1 (lanes 6-10), were electroporated using voltages of 120-190 V as indicated in the presence of 66 or 33µCi [α32P]GTP, respectively. Proteins were extracted, and the Rasbound GTP and GDP were separated as described earlier. As a control (lane 11), 2H1 cells growing in a 3cm petri to were metabolically labelled with 200 µCi [α32P] orthophosphate and processed as described previously. (C) Assessment of Ras activity in normal 10T½ fibroblasts and their ras-transformed counterparts, 2H1, using the standard SLO permeabilization assay. This assay was performed as described (Brownell et al., 1997) and is shown here as a comparison. Lane 1, 10T½; lane 2, rasval12-transformed, 10T½-derived line 2H1; and lane 3, 2H1 precipitated with control rat IgG instead of anti-Ras antibodies. |
Solutions
- [γ32P]ATP: 600-1000µCi/ml in phosphate-free DMEM.
- Simian virus large tumor antigen (SVLT) or adenovirus E1A extraction buffer: 122 mM NaCl, 18 mM Tris-base, pH 9.0, 0.8 mM CaCl2, 0.43 mM MgCl2, 10% glycerol, 1% NP-40, 10µg/ml aprotinin, 10µg/ml leupeptin, 1 mM PMSF, and 1 mM vanadate. Add 8.0g NaCl, 2.42 g Tris-base, 0.1 g CaCl2, 0.04 g MgCl2, 10 ml NP-40, and 100ml glycerol to 800ml H2O. Adjust pH to 9.0 and store at -20°C. On the day of the experiment, add 0.1 ml aprotinin stock, 0.1 ml leupeptin stock, 1 ml PMSF stock, and 0.1 ml vanadate stock solutions to 100 ml of this solution.
- Tris-LiCl solution for washing immunoprecipitates: 0.1M Tris-base and 0.5M LiCl. Make a stock solution of 10X (4 litres). Add 484g of Tris-base and 848 g of LiCl in 4 litres of H2O. Adjust pH to 7.0 using concentrated HCl.
- SDS gel-loading buffer: 15ml 10% SDS, 1.5ml mercaptoethanol, 6ml glycerol, 3ml 1.25M Tris, pH 6.8, 0.75 ml 0.3% bromphenol blue, and water to 30ml.
- 10% SDS stock solution
- 1.25M Tris-HCl (pH 6.8) stock solution: Add 151.3g Tris-base to 800ml distilled water. Adjust the pH to 6.8 using concentrated HCl and the volume to 1 liter with distilled water.
- Plate, starve from phosphates, and wash the cells with phosphate-free DMEM as in Section III,A.
- Add the [γ32P]ATP solution.
- Apply three to six pulses of the appropriate strength (40-200 V, see Comment 3) from a 10- or 20-µF capacitor, depending on the apparatus used (Fig. 1A vs Fig. 1C). If the setup in Fig. 1 A is employed, then the solution can be carefully aspirated and reused once more after the pulse.
- Add phosphate-free medium and incubate the cells for 2-3 h in a tissue-culture incubator.
- Extract the proteins by adding 1 ml SVLT extraction buffer to the window, scraping into a 15 ml tube, and rocking on ice. Clarify by spinning for 30min at 1,000rpm in a Beckman J-6 centrifuge (2,000g).
- Precipitate with the pAbl08 (SVLT) or M73 (E1A) monoclonal antibody and wash three times with PBS, twice with the Tris-LiCl solution, and once with H2O. Elute labelled proteins from the beads with SDS geMoading buffer and resolve by acrylamide gel electrophoresis (Fig. 3).
![FIGURE 3 Labeling of the simian virus 40 large tumor antigen or adenovirus EIA through in situ electroporation
of [γ32P]ATP. (A) Mouse 10T½ cells (lane 1) or their SVLT-transformed derivatives (line 10SV2b, lanes 2-8) were grown on 50 × 30mm conductive areas (Figs. 1C and 1D). A solution containing 50µCi [γ32P]ATP in phosphate-free DMEM was added to the cells, and six capacitor-discharge pulses of 10µE 40-80 V were applied as indicated. Cells were placed in a humidified incubator for 30min. For a comparison (lanes 9 and 10), the same SVLT-transformed cells were labeled in vivo with the indicated amounts of [32P]orthophosphate. SVLT was precipitated from detergent extracts with the pAbl08, anti-SVLT antibody (lanes 1, 3-7, and 10), or normal mouse IgG (lanes 2, 8, and 9) and labeled proteins were resolved by acrylamide gel electrophoresis. Dried gels were exposed for 1 h to Kodak XAR-5 film with an intensifying screen. Note the intense and specific labeling of SVLT and the associated phosphoprotein, p53, by in situ electroporation (lanes 5 and 6) compared to cells labeled in vivo with 200µCi [32P]orthophosphate (lanes 9 and 10). (B) Human 293 cells transformed with adenovirus DNA were labeled as described earlier and extracts were preadsorbed with normal mouse IgG (lanes 2, 4, 6, 8, and 10) or immunoprecipitated using the M73, anti-E1A antibody (lanes 1, 3, 5, 7, and 9). Bracket points to the position of the phosphorylated E1A bands. M, molecular weight marker lanes.](images/v2_pb_s08_c43_f03.jpg) |
| FIGURE 3 Labeling of the simian virus 40 large tumor antigen or adenovirus EIA through in situ electroporation of [γ32P]ATP. (A) Mouse 10T½ cells (lane 1) or their SVLT-transformed derivatives (line 10SV2b, lanes 2-8) were grown on 50 × 30mm conductive areas (Figs. 1C and 1D). A solution containing 50µCi [γ32P]ATP in phosphate-free DMEM was added to the cells, and six capacitor-discharge pulses of 10µE 40-80 V were applied as indicated. Cells were placed in a humidified incubator for 30min. For a comparison (lanes 9 and 10), the same SVLT-transformed cells were labeled in vivo with the indicated amounts of [32P]orthophosphate. SVLT was precipitated from detergent extracts with the pAbl08, anti-SVLT antibody (lanes 1, 3-7, and 10), or normal mouse IgG (lanes 2, 8, and 9) and labeled proteins were resolved by acrylamide gel electrophoresis. Dried gels were exposed for 1 h to Kodak XAR-5 film with an intensifying screen. Note the intense and specific labeling of SVLT and the associated phosphoprotein, p53, by in situ electroporation (lanes 5 and 6) compared to cells labeled in vivo with 200µCi [32P]orthophosphate (lanes 9 and 10). (B) Human 293 cells transformed with adenovirus DNA were labeled as described earlier and extracts were preadsorbed with normal mouse IgG (lanes 2, 4, 6, 8, and 10) or immunoprecipitated using the M73, anti-E1A antibody (lanes 1, 3, 5, 7, and 9). Bracket points to the position of the phosphorylated E1A bands. M, molecular weight marker lanes. |
The technique of in situ electroporation is very versatile. A large variety of molecules, such as peptides (Boccaccio et al., 1998; Raptis et al., 2000), nucleotides (Brownell et al., 1997), antibodies (Raptis and Firth, 1990), or drugs (Marais et al., 1997), can be introduced, alone or in combination, at the same or different times, in continuously growing or growth arrested cells or cells at different stages of thefir division cycle (see chapter 44 by Raptis et al.).
- The slides can normally be washed with Extran- 300 and reused a number of times. However, in the case of introduction of radioactive material, exposure of personnel to irradiation is an important consideration. The removal of 32P-labelled nucleotides from the ITO-coated glass is difficult because the phosphate group is attracted to this coating (Tomai et al., 2000), hence the use of inexpensive slides and electrodes that can be discarded after use is highly desirable. Slides with a conductivity of 20Ω/sq are sufficiently inexpensive that they can be discarded after use or stored for the 32P to decay before washing. The use of less conductive ITO-coated glass (100Ω/sq) would reduce the cost of the slides further. However, in our experience, the conductivity of this grade of glass is not sufficiently consistent for electroporation experiments due to problems related to uniformity of thickness encountered with the thinner coating of the less conductive, commercially available surface. On the other hand, the use of more conductive glass (2Ω/sq) requires a smaller electrode angle, which reduces the cost of the material substantially. However, this glass is not a regular production item, hence it is more expensive.
- The [α32P]GTP must be of the highest purity. Because a number of lots were found to contain varying amounts of [α32P]GDP, it is wise to test the preparation by thin-layer chromatography before use.
- Determination of the optimal voltage and capacitance. Electrical field strength has been shown to be a critical parameter for cell permeation, as well as viability (Chang et al., 1992). It is generally easier to select a discrete capacitance value and then control the voltage precisely. The optimal voltage depends on the strain and metabolic state of the cells, as well as the degree of cell contact with the conductive surface, possibly due to the larger amounts of current passing through an extended cell (Yang et al., 1995; Raptis and Firth, 1990). Densely growing, transformed cells or cells in a clump require higher voltages for optimum permeation than sparse, subconfluent cells. Similarly, cells that have been detached from their growth surface by vigorous pipetting prior to electroporation require substantially higher voltages. In addition, cells growing and electroporated on collagen, poly-L-lysine, or CelTakTM-coated slides require substantially higher voltages than cells growing directly on the slide.
![FIGURE 4 Effect of field strength on the introduction of nucleotides. Six pulses of increasing voltage were applied from a 10µF capacitor to serum-starved 10T½ cells growing on a conductive surface of 50 × 30mm in the presence of 10µCi [α32P]GTP () or 10µCi [γ32P]ATP (). Total protein labelling was quantitated using a nitrocellulose filter-binding assay (Buday and Downward, 1993). Numbers refer to cpm per 100µg of protein in clarified extracts. As a control, cells were electroporated with 5 mg/ml Lucifer yellow (O) and its introduction was assessed by fluorescence measurement of cell lysates using a Perkin-Elmer Model 204A fluorescence spectrophotometer. Cell killing () was calculated from the plating efficiency of the cells 2h after the pulse [from Raptis et al. (2003) and Tomai et al. (2003), reprinted with permission].](images/v2_pb_s08_c43_f04.jpg) |
| FIGURE 4 Effect of field strength on the introduction of nucleotides. Six pulses of increasing voltage were applied from a 10µF capacitor to serum-starved 10T½ cells growing on a conductive surface of 50 × 30mm in the presence of 10µCi [α32P]GTP ( |
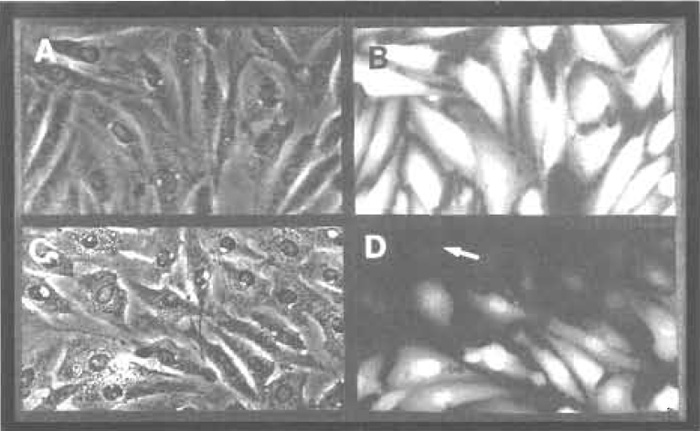 |
| FIGURE 5 Determination of optimal voltage. Rat Flll fibroblasts growing on conductive slides (Figs. 1A and 1B, cell growth area, 32 × 10mm) were electroporated in the presence of 5mg/ml Lucifer yellow using three pulses of 155 (A and B) or 190 (C and D) volts delivered from a 20µF capacitor. After washing of the unincorporated dye, cells were photographed under phase-contrast (A and C) or fluorescence (B and D) illumination. Arrows in C and D point to a cell that has been killed by the pulse. Note the dark, pycnotic and prominent nucleus under phase contrast and the fiat, nonrefractile appearance. Such cells do not retain any electroporated material, as shown by the absence of fluorescence (C). It is especially striking that cells at the top of frames C and D, situated at the edge of the electroporated area, received a larger amount of current and have been killed by the pulse. Magnification: 240 times. |
Prepare a series of slides with cells plated uniformly in a 32 × 10 mm window (Figs. 1A and 1B). Set the apparatus at 20µF capacitance. Prepare a solution of 5 mg/ml Lucifer yellow in phosphate-free DMEM and a solution of [α32P]GTP or [γ32P]ATP containing 5 mg/ml Lucifer yellow in phosphate-free DMEM. Electroporate the Lucifer yellow solution at different voltages, 20µF, three pulses, to determine the upper limits where a small fraction of the cells at the corners of the window (usually the more extended ones) are killed by the pulse, as determined by visual examination under phase-contrast and fluorescence illumination 5-10 min after the pulse (Fig. 5). Depending on the cells and growth conditions, this voltage can vary from 130 to 190 V. Repeat the electroporation using the radioactive nucleotide solution at different voltages starting at 20 V below the upper limit and at 5V increments. The Lucifer yellow offers a convenient marker for cell permeation and it was found not to affect the results.
- Serum was shown to facilitate pore closure (Bahnson and Boggs, 1990).
- The slides come with the apparatus and are sterile. If sterility is compromised or if the slides have been washed to reuse, then place in petris and sterilize by adding 80% ethanol for 20min and then removing the ethanol by rinsing with sterile distilled water.
Measurement of steady-state Ras activity is possible using this method because, contrary to other methods of cell permeabilization, such as streptolysin-O (Buday and Downward, 1993), the cells are not detectably affected by the procedure so that they can be incubated for long periods of time before extraction. In addition, electroporation does not appear to induce a rapid breakdown of intracellular GTP in any of the lines tested, even under conditions where a substantial fraction of the cells are killed by the pulse (Fig. 2). As a result, determination of the Ras-bound, GTP/GTP + GDP ratio is made easier by the fact that although the optimal voltage must be determined empirically as in all electroporation experiments, excessively high voltages, despite the fact that they may kill a substantial proportion of the cells, do not alter the ratios obtained (Fig. 2B), presumably because such cells rapidly lyse, without affecting the results.
- Care must be taken so that cells do not dry during the procedure, especially during wiping of the frame with a tightly folded Kleenex. It was found that serum-starved cells were especially susceptible. The morphology of cells that have been killed by drying is very similar to cells that have been killed by the pulse (Fig. 5).
- Accurate determination of the optimal voltage is very important. For most nucleotide introduction applications, optimal labelling was observed in the range of 140-160 V, whereas these margins were found to be narrower for serum-starved cells. Nevertheless, the Ras-bound GTP/GTP + GDP ratios or the profile of SVLT or EIA labelling obtained was found to be the same even when a substantial proportion of the cells were killed by the pulse, in which case merely 32P incorporation is reduced.
Acknowledgments
The financial assistance of the Canadian Institutes of Health Research, the Natural Sciences and Engineering Research Council of Canada, and the Cancer Research Society Inc. to LR is gratefully acknowledged. AV is the recipient of NSERC and Ontario Graduate studentships, a Queen's University graduate award, and a Queen's University travel grant. ET is the recipient of a Queen's University graduate award, an NSERC studentship, and awards from the Thoracic Society and the Lemos foundation. HB was the recipient of a studentship from the Medical Research Council of Canada and Queen's University graduate school and Microbix Biosystems Inc. travel awards.
Bahnson, A. B., and Boggs, S. S. (1990). Addition of serum to electroporated cells enhances survival and transfection efficiency. Biochem. Biophys. Res. Commun. 171, 752-757.
Boccaccio, C., Ando, M., Tamagnone, L., Bardelli, A., Michielli, P., Battistini, C., and Comoglio, P. M. (1998). Induction of epithelial tubules by growth factor HGF depends on the STAT pathway. Nature 391, 285-288.
Brownell, H. L., Firth, K. L., Kawauchi, K., Delovitch, T. L., and Raptis, L. (1997). A novel technique for the study of Ras activation; electroporation of [α32P]GTP. DNA Cell Biol. 16, 103-110.
Brownell, H. L., Lydon, N., Schaefer, E., Roberts, T. M., and Raptis, L. (1998). Inhibition of epidermal growth factor-mediated ERK½ activation by in situ electroporation of nonpermeant [(alkylamino)methyl]acrylophenone derivatives. DNA Cell Biol. 17, 265-274.
Brownell, H. L., Narsimhan, R., Corbley, M. J., Mann, V. M., Whitfield, J. E, and Raptis, L. (1996). Ras is involved in gap junction closure in mouse fibroblasts or preadipocytes but not in differentiated adipocytes. DNA Cell Biol. 15, 443-451.
Chang, D. C., Chassy, B. M., Saunders, J. A., and Sowers, A. E. (1992). Guide to Electroporation and Electrofusion. Academic Press, New York.
Downward, J. (1995). Measurement of nucleotide exchange and hydrolysis activities in immunoprecipitates. Methods Enzymol. 255, 110-117.
Egawa, K., Sharma, P. M., Nakashima, M., Huang, Y., Huver, E., Boss, G. R., and Olefsky, J. M. (1999). Membrane-targeted phosphatidylinositol kinase mimics insulin actions and induces a state of cellular insulin resistance. J. Biol. Chem. 274, 14306-14314.
Lowy, D. R., and Willumsen, B. M. (1993). Function and regulation of ras. Annu. Rev. Biochem. 62, 851-891.
Raptis, L., Brownell, H. L., Vultur, A. M., Ross, G., Tremblay, E., and Elliott, B. E. (2000). Specific inhibition of growth factorstimulated ERK½ activation in intact cells by electroporation of a Grb2-SH2 binding peptide. Cell Growth Differ. 11, 293-303.
Raptis, L., Brownell, H. L., Wood, K., Corbley, M., Wang, D., and Haliotis, T. (1997). Cellular ras gene activity is required for full neoplastic transformation by simian virus 40. Cell Growth Differ. 8, 891-901.
Raptis, L., and Firth, K. L. (1990). Electroporation of adherent cells in situ. DNA Cell Biol. 9, 615-621.
Raptis, L., Vultur, A., Balboa, V., Hsu, T., Turkson, J., Jove, R., and Firth, K. L. (2003). In situ electroporation of large numbers of cells using minimal volumes of material. Anal. Biochemi.
Robinson, M. J., and Cobb, M. H. (1997). Mitogen-activated protein kinase pathways. Curr. Opini. Cell Biol. 9, 180-186.
Scheele, J. S., Rhee, J. M., and Boss, G. R. (1995). Determination of absolute amounts of GDP and GTP bound to Ras in mammalian cells: Comparison of parental and Ras-overproducing NIH 3T3 fibroblasts. Proc. Natl. Acad. Sci. USA 92, 1097-1100.
Tomai, E., Klein, S., Firth, K. L., and Raptis, L. (2000). Growth on indium-tin oxide-coated glass enhances 32P-phosphate uptake and protein labelling of adherent cells. Prep. Biochem. Biotechnol. 30, 313-320.
Tomai, E., Vultur, A., Balboa, V., Hsu, T., Brownell, H. L., Firth, K. L., and Raptis, L. (2003). In situ electroporation of radioactive compounds into adherent cells.
Yang, T. A., Heiser, W. C., and Sedivy, J. M. (1995). Efficient in situ electroporation of mammalian cells grown on microporous membranes. Nucleic Acids. Res. 23, 2803-2810.
Support our developers




