Separation of Cell Populations Synchronized in Cell Cycle Phase by Centrifugal Elutriation
Centrifugal elutriation is the only method whereby large numbers of cells can be separated rapidly on the basis of size (Diamond, 1991; Merrill, 1998; Davis et al., 2001). The capability of discriminating between very small differences in cell size provides the ability to separate cells into sequential cell cycle phase populations of relatively high purity without the use of drugs or inhibitors (Bludau et al., 1986; Braunstein et al., 1982; Hann et al., 1985; Iqbal et al., 1984; Wu et al., 1993; Brown and Schildkraut, 1979; Bialkowski and Kasprzak, 2000; Datta and Long, 2002; Deacon et al., 2002; Hengstschlager et al., 1999; Houser et al., 2001; Karas et al., 1999; Rehak et al., 2000; Sugikawa et al., 1999; Syljuasen and McBride, 1999; Van Leeuwen-Stok et al., 1998). It has been used successfully to separate a wide variety of cell types from suspension and substrate-dependent cultures and to separate mixed cell populations liberated directly from tissues or body fluids (Boerma et al., 2002; Dagher et al., 2002; Wong et al., 2001, 2002). The purity of the samples is relatively high and the cells proceed to grow, following separation, without a detectable lag period. Thus, centrifugal elutriation combines speed of separation of large numbers of cells with little or no perturbation of the cell growth cycle and avoids the use of agents that might induce artifact. As additional advantages, centrifugal elutriation overcomes the limits on cell number imposed by fluorescence-activated cell sorting and the long separation times required for unit gravity sedimentation, as well as problems associated with osmotic stress in centrifugation media. The only real compromise is that the purity of the samples is somewhat lower than commonly achieved with alternative methods. The developmental history and theory of centrifugal elutriation are reviewed elsewhere (Conkie, 1985; Beckman Instruments, 1990).
Centrifugal elutriation imposes two opposing forces on mixed cell populations to facilitate their fractionation into subpopulations (Fig. 1 and see Section IV). This technology has proven to be effective in fractionating cells, based on very small differences in cell size, with nominal cross contamination, and in numbers unmatched by other methods of cell separation. In addition, centrifugal elutriation can be performed rapidly, requiring only a few minutes (usually 20- 120min) to affect separation and this occurs in media containing no special additives that might affect the osmolarity or viscosity of the medium. Thus, with very little physiological change perceived by the cells, separation can be affected rapidly. Some shear force is exerted on the cells during separation, but this is not sufficient to appreciably affect viability or behavior of the cells in most cases. Compared to other methods of separation, such as fluorescence-activated cell sorting or unit gravity separation, centrifugal elutriation is by far the most gentle and the most rapid in manipulation and separation of the cell populations.
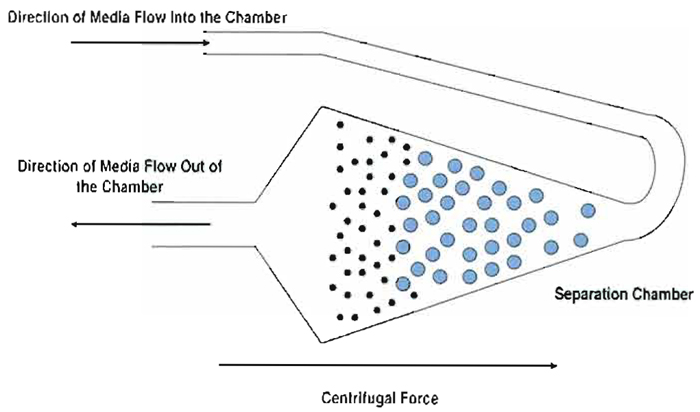 |
| FIGURE 1 Cell separation dynamics and opposing forces in the elutriation chamber. The flow of media is opposed by centrifugal force in a balance that holds particles in equilibrium in the separation chamber. Due to different levels of force applied proportional to surface area presented, smallest particles sort to the inside of the chamber relative to larger particles. Separation is achieved by increasing the flow rate incrementally to push the smallest population of particles past the widest part of the chamber and into the inner narrowing section where media flow accelerates affecting elutriation of the population. |
II. MATERIALS AND INSTRUMENTATION
The centrifugal elutriator is from Beckman Instruments (elutriator rotor assembly Cat. No. JE-6B was run in a Model J2-21 elutriation centrifuge). Accessories are used as specified by the manufacturer throughou t, and the rotor is equipped with a standard separation chamber. A Masterflex digital peristaltic pump (0-100rpm Model 07523-70 fitted with a model 7014-21 pump head, Cole-Parmer) is used to pump cells and media through the rotor.
III. PROCEDURES
A. Cell Culture
Growth medium and elutriation medium are used per standard protocols for the growth of HeLa cells (Pai and Bird, 1992). If different cell lines are to be elutriated, appropriate media should be substituted (see Section V).
- Growth and elutriation media: Dissolve powdered α-MEM (10-liter pack) in ultrapure water containing a final concentration of 1× antibiotics and 22.2 g NaHCO3 and make up to a total volume of 10 liters. Place the medium in a pressurizable vessel (Millipore), filter through a 0.2-µm filter (Corning) by positive pressure into sterile 0.5-liter bottles, and store at 4°C. Prior to use, add FBS (10%, v/v) to make growth medium or add DHS (5%, v/v) to make elutriation medium. Use of DHS reduces the cost of elutriation greatly without detectable effects on cell growth or quality of the fractionation (see Section V).
- 0.5M HEPES: Dissolve 14.17g HEPES buffer (Fisher, Cat. No. BP310-100) in ~80 ml water and adjust pH to 7.2. Adjust volume to 100 ml, filter sterilize, and store in the cold at 4°C.
- 0.5M Na2EDTA: Begin to dissolve 14.61 g EDTAfree acid (Fisher, Cat. No. BPl18-500) in ~40ml water with a magnetic stir bar. Monitor pH of the solution continuously while slowly adding 1M NaOH (40g/ liter for 1M stock) dropwise. Continue to adjust the pH up to ~8.0. As the EDTA dissolves, the pH will continue to fall. Carefully adjust the final pH to 8.0 without exceeding this value. Adjust volume to 100 ml, filter sterilize, and store at 4°C.
- Trypsin solution: Add 8ml of 2.5% trypsin stock solution (10 ×), 2 ml 0.5M HEPES, pH 7.2, and 2 ml 0.5M Na2EDTA, pH 8.0, to 100ml of Hanks' salt solution. Make additions from sterile stock solutions, maintaining sterility of the final solution. Store at 4°C.
- 70% ethanol: Combine 700ml absolute (not denatured) ethanol with 300ml water. Store in 0.5-liter bottles at room temperature.
Steps
- Grow cultured HeLa S3 cells in 20 ml of modified Eagle's α-MEM medium (Invitrogen/Gibco) with 10% FBS and antibiotics in a 15-cm diameter culture plates at 37°C with 5% CO2 and 100% humidity (Pai and Bird, 1992).
- Collect cells at 60-70% confluence by trypsin digestion. Harvest just before the rotor is filled with medium.
- Concentrate cells by low-speed centrifugation (3000g for 5 min) at 4°C and resuspend in 5 ml of ice cold α-MEM with 5% DHS for every three plates (15cm). Use this medium throughout the elutriation procedure. Maintain the cells on ice until they are loaded into the elutriator.
B. Elutriation
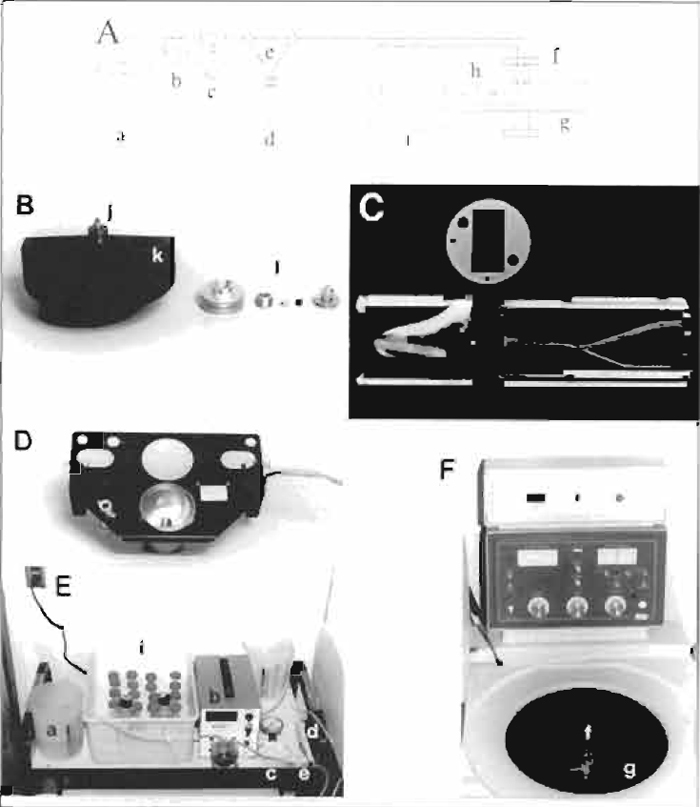 |
| FIGURE 2 Assembly and setup of centrifugal elutriator. (A) Schematic of the centrifugal elutriator. Elutriation medium is pumped from the reservoir beaker (a) by the peristaltic pump (b) through the pressure gauge (c) and the sample tube (d) or the bypass harness (e) through the rotating seal (f) and into the rotor (g). The sample is separated in the elutriation chamber (h) and is pumped back to the sample collection tubes on ice (i). (B) Rotor and rotating seal assembly, including O rings on top of the rotor (j), which seal the rotating assembly to the rotor, the elutriation chamber (k), and, from left to right, the outer ring, lower washer, the spring that is placed inside the lower washer, rotating seal, and top of the seal assembly (1). (C) Elutriation chamber showing left (outer) and right (inner) halves separated by the gasket (above). Note the actual separation chamber within the left side and the set screws and alignment pins extending beyond both edges of the right side of the chamber. (D) Strobe assembly located below the rotor. (E) Elutriation setup showing the reservoir beaker (a), the peristaltic pump (b), pressure gauge (c), sample tube (d), bypass harness (e), and sample collection tubes (i). Note the positions of beakers to supply/accept media. (F) Elutriation centrifuge showing the complete rotating seal assembly (f), elutriation rotor (g), with strobe wires/waste tubing extending through the right centrifuge wall, and the inlet/outlet tubing extending through the left centrifuge wall. |
- Arrange the elutriation system as described in the schematic diagram (Fig. 2A). Assemble the elutriator rotor according to the manufacturer's directions (Fig. 2B). Lightly lubricate each O ring, which seals the rotating assembly on top of the rotor, with silicone grease, taking care to wipe off any excess. Place the lower washer and spring on top of the rotor followed by the outer ring and rotating seal. Note that the screw threads on the top assembly are reversed. Lightly tighten the top and check that the outer ring spins freely. Tighten the side set screw and repeat the check for a freely spinning assembly. Loosen the lower washer inside the outer ring half a turn. Check that the assembly spins freely, retighten the lower washer, and check that it spins freely again. Care should be taken to ensure that the rotating seal connecting the rotor to the fluid lines is freshly cleaned and polished with the scintered glass plate and polishing paper provided. Only a lint-free tissue with an appropriate solvent (e.g., CHCl3) should be used as even a small speck of lint can cause leakage. A very thin layer of silicone grease can be applied to the upper edge of the rotating seal to help create and maintain a good seal but all excess silicone must be removed. Stick the seal to the polypropylene disk on the bottom surface of the top of the seal assembly by gently seating the silicone on the seal with a half turn. Carefully screw the top of the seal assembly onto the outer ring (note reverse threads) and hand tighten. This connection should not leak more than 1-3 ml during a normal elutriation run of <1 h. Even very small leaks encountered at low initial flow rates can result in significant loss of fluid and cells toward the end of the run as the flow rate rises. If perpersistent leaks are encountered, reexamine the seal and check for a perfectly clean and even surface on both edges. Repolish the rotating seal if necessary and then repeat the assembly procedure. If none of these measures seals the rotor connections adequately, the seal should be replaced.
- Assemble the elutriator chamber according to the manufacturer's directions (Fig. 2B). Ensure that both halves of the chamber are perfectly clean and that the polypropylene gasket is positioned to allow alignment of the sample tube (see Section V). Tighten the screws to assemble but do not overtighten. Lightly lubricate both O rings, on the base of the chamber assembly, with silicone grease, taking care to wipe off any excess. Insert the chamber and align the base pin and sample tube connections. Secure it in place with the metal plug. The screw threads on the plug should be lubricated with Spincoat (Beckman) provided with the elutriator. Tighten with the tool provided but do not overtighten as the O rings can be crushed.
- Assemble the elutriator centrifuge according to the manufacturers directions (Fig. 2C). Remove the high-speed rotor (if present) from the centrifuge and wipe out any moisture in the centrifuge chamber. Install the strobe assembly in the centrifuge chamber and secure with thumb screws while ensuring that it is centered over the rotor spindle in the center (Fig. 2D). Feed both wire connections through the ports on the right side of the centrifuge chamber and secure them with the metal plate. Carefully place the rotor on the spindle in the center of the centrifuge and ensure that it spins freely. Connect the three pieces of silicone tubing to the rotor (inlet, outlet, and overflow) and feed them through the appropriate port (inlet and outlet to the left, overflow to the right with the wires). Ensure that all are pulled tight enough so that none have any slack in them but not so tight as to pinch or pull off any of the connections (Fig. 2F). The overflow tube should wrap around the top of the seal assembly and pass under the inlet port (the upper of the two ports on the sides of the top of the seal assembly). Seal each of the ports around each tube and wire with a slit stopper (provided with the centrifuge), where they pass through the centrifuge chamber wall, to ensure a good vacuum.
- Prepare a large ice bucket containing two 0.5-liter bottles of elutriation medium, 30 sterile-capped tubes (50ml), and the cell suspension (Fig. 2E). Place the bucket on a cart or table next to the centrifuge with the pump and three 2-liter beakers. One beaker contains 1 liter of sterile water. Include a 20-ml syringe with an 18-gauge needle and the sample injection harness with a pressure gauge (Cole-Parmer, Cat. No. EW-07380-75). If sterile samples are to be collected, an additional beaker of 70% ethanol and a bottle of sterile water must also be included. If elutriated cells are to be cultured for more than a few hours after separation, a sterile hood must be positioned next to the centrifuge to allow both reservoir beakers and samples to be collected under sterile conditions.
- Clean the exterior of the silicone tubing with ethanol and place the inlet in the beaker containing the sterile water and the outlet in an empty beaker. The inlet tubing should also be installed into the pump head and attached to the sample application and bypass harness, with an inline pressure gauge, so that they are between the pump and the rotor (Fig. 2E). Begin pumping water through the rotor (which is stationary and the centrifuge is off, see Section V) at 45ml/min (>200ml). Carefully observe the water as it fills the equipment (~100ml) and dislodge any bubbles with gentle pressure on the tubing or tapping of other components. Release the air from dead spaces within the pressure gauge by pinching the tubing just after the gauge and releasing it rapidly. Do not let the pressure rise above 15psi or the tubing may burst. Adjust the stopcocks on the sample/bypass harness to allow the harness, stopcocks, and sample tube to fill completely. Continue to pump water through the equipment until all the bubbles have been cleared. Observe all connections, particularly at the rotor, for leakage.
- Close the centrifuge door and start the rotor. Allow the speed to gradually rise and stabilize at 2000rpm (±1rpm is acceptable) at 4°C. Be sure to increase the speed slowly as the fine speed control can easily overshoot the set point on acceleration. Check each of the stoppers to ensure that the seals are adequate to allow development of a vacuum in the centrifuge. Observe the chamber through the periscope assembly in the door and adjust the strobe firing timer to center the image. If the chamber appears to have a rod running along the center rather than two screws running near each edge, then the strobe is 180° out of phase with the rotor and is allowing visualization of the balance chamber, not the elutriation chamber. Continue to adjust the strobe until the elutriation chamber is visible. Check for bubbles at the outlet using the squeeze-and-release technique described earlier. Turn off the pump and observe the outlet tubing as you raise it out of the beaker and suspend it free in the air. If a slow leak occurs, the fluid and air will be drawn back up into the tube as the fluid leaks out of the system. The system is sealed if the fluid level in the outlet tube does not change.
- If sterile collection is required, switch to 70% ethanol and pump 200ml through the rotor followed by 200ml of sterile water from a bottle in the laminar flow hood. Switch to elutriation medium (without DHS) on ice and pump 200ml (if sterile collection is not required, omit ethanol rinse). Be sure to include all sections of the sample injection harness, including the sample tube and the stopcocks, during the rinse with each of these solutions. Turn off the pump before switching the stopcocks. Turn the pump off and change to elutriation medium with 5 % DHS to prevent the cells from sticking. Pump 100ml at 45ml/min through the sample tube. At this point, two people are required to operate the system: one collects samples and one monitors rotor and pump speeds as well as managing sample injection.
- Disperse the trypsinized cells (~1-2 × 108), which have been held on ice in 5 ml of elutriation medium (containing 5% DHS), with seven very gentle passes through an 18-gauge needle on a 20-ml syringe. Be careful not to introduce bubbles. Turn off the pump. Gently inject the cells into the sample tube (positioned with the stopper up), allowing the cells to settle to the bottom of the tube. Carefully turn the tube over (stopper down) and gently inject about 2 ml of air into the sample tube to act as a shock absorber against the pulsing peristaltic action of the pump. Adjust the pump, which is still off, to zero and then turn the pump on. Gradually adjust the pump up to 10 ml/min, being careful not to overshoot this value. Observe the sample tube and watch the cells enter the system. Take care to avoid pulsing of the medium or leaving a residual pools of cells in the sample tube. Collect three tubes of 50ml each in the sample collection tubes on ice. Once the cells are loaded, the harness can be set on the bench in a stable position that maintains the inverted orientation of the sample tube (stopper down). Continue to monitor the entry of the cells into the elutriation chamber through the periscope. Carefully increase the pump speed to 15 ml/min. Do not overshoot. We collect G1-phase cells between 21 and 25ml/min, Sphase cells at 29-35ml/min, and G2/M-phase cells at 43 ml/min. Be prepared to work quickly at the end of an elutriation run as the flow rate becomes very fast and tubes containing elutriated cells fill at the rate of approximately one every minute. Following elutriation, pellet the cells by centrifugation (3000g 5min) and resuspend in growth medium. Adjust cell density with additional growth medium, after determination of cell concentration in each sample by counting in a hemacytometer, and transfer to tissue culture dishes.
C. Cell Cycle Analysis
Part of the synchronous cell fractions are fixed and analyzed by flow cytometry and the remainder of the cells are immediately prepared for RNA isolation or placed back into culture for further manipulation (Fig. 3). Cultured cells are analyzed for their ability to synchronously enter S phase by determining the kinetics of [3H]thymidine incorporation (Fig. 4).
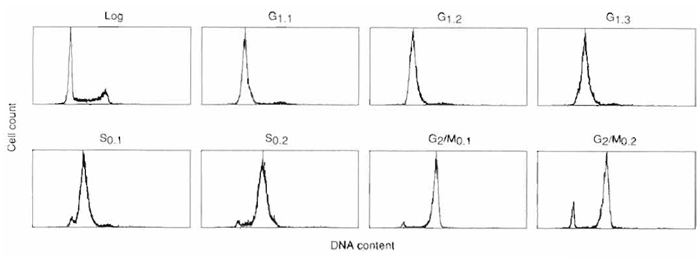 |
| FIGURE 3 Flow cytometric analysis of cell cycle synchrony in sequential centrifugal elutriator fractions of HeLa S3 cells. Synchronous populations of HeLa S3 cells were selected by centrifugal elutriation of exponential cultures (log), and the degree of synchrony was analyzed by flow cytometry. Cell number was plotted against DNA content based on propidium iodide fluorescence for each cell cycle fraction. Flow cytometric analysis of sequential G1-phase fractions collected at flow rates of 21ml/min (G1.1), 23ml/min (G1.2), and 25ml/min (G1.3). S-phase cells were selected by centrifugal elutriation at flow rates of 29ml/min (S0.1) and 35ml/min (S0.2), and G2/M-phase cells were collected at 43 ml/min. |
- Phosphate-buffered saline (PBS): Dissolve 0.71 g of Na2HPO4 (0.01M final, Sigma, Cat. No. S-1934) and 4.5g NaCl (0.9%, w/v final) in ~400ml water and adjust pH to 7.6. Adjust volume to 500 ml, filter sterilize, and store at 4°C.
- Staining solution: Make 50µg/ml propidium iodide (PI) and 40 µg/ml RNase A by dilution of stock solute/ons. Add 111µl PI stock (4.5mg/ml, Sigma, Cat. No. P-1764) and 20µl RNase A stock (20mg/ml, Sigma, Cat. No. R-1859) to 10ml water. Do not attempt to weigh RNase as any contamination will make future isolation of RNA very difficult. Open a 250-mg bottle in the fume hood and add 12.5 ml water. Cap, dissolve, and boil for 15min to inactivate DNase (Sambrook et al., 1989). Aliquot with a plugged disposable pipette (tissue culture type) in 1-ml lots in microcentrifuge tubes. Store at approximately -20°C. Use only disposable tubes and pipettes with RNase solutions and avoid any contamination or aerosols. Dissolve 100mg of PI in 22.22 ml of water directly in the bottle. Do not weigh out. PI is extremely toxic and is a carcinogen as well as a potential mutagen and should be handled with care, including the use of gloves. Be careful not to create aerosols or liberate dust from the granular reagent. Dispose of all PI solutions and contaminated materials as hazardous waste. Use only disposable tubes and pipettes. Store PI at -20°C in the dark as it is light sensitive.
- 100% trichloroacetic acid: To a 500g bottle of trichloroacetic acid (TCA, Sigma, Cat.tNo. T-2069), add sufficient water to bring the volumel in the bottle to approximately the shoulder. Add a stir bar, cap the bottle, and stir to dissolve the contents. Carefully adjust the volume to 500ml. TCA is extremely caustic. Do not attempt to measure or weigh TCA granules. Use caution when pipetting the solution. TCA 100% solution is very stable when stored at approximately ~4°C. Dilutions should be freshly prepared from the stock daily. Store dilutions on ice while in use and dispose of unused portions.
- [3H]Thymidine growth medium: Add 10µCi/µl [3H]thymidine (1 µCi/µl stock, PerkinElmer Life and Analytical Sciences, Inc., Cat. No. NET-027Z) directly to growth medium under sterile conditions at the rate of 10 µl/ml of medium. Prepare fresh on a daily basis.
- 1.0M Tris buffer: Dissolve 60.55g Tris buffer (Fisher, Cat. No. BP152-1) in ~400ml water and adjust pH to 7.6. Adjust volume to 500 ml, filter sterilize, and store at 4°C.
- 20% sodium dodecyl sulfate (SDS): Dissolve 20g SDS (Sigma, Cat. No. L-1926) in sterile water using a sterile beaker and a stir bar rinsed in ethanol. Adjust final volume to 100ml. SDS cannot be autoclaved or filtered. Store at room temperature.
- TES buffer: Add 10mM Tris-HCl, pH 7.6 (1 ml of 1M stock), 1 mM EDTA, pH 8.0 (0.5 ml of 0.5 M stock), and 1% SDS (5 ml of 20% stock) to 93.5 ml water, filter, and store at room temperature.
- 4% paraformaldehyde: Dissolve 4 g paraformaldehyde (Electron Microscopy Sciences, Cat. No. 15710) in 100ml water and stir to dissolve by heating gently to 60-65°C in a fume hood. This can take an extended period and the solution will still appear cloudy. Clarify by the addition of a few drops of 1M NaOH (up to ~20 drops). Cool before use and store at 4°C. Fixative fumes are extremely toxic. Always use in a fume hood and avoid any contact.
![FIGURE 4 Analysis of synchrony of entry into DNA synthesis in centrifugal elutriation fractions of HeLa S3 cells. HeLa cells were separated into synchronous fractions by centrifugal elutriation, and kinetics of entry into DNA synthesis phase (S phase) was determined by measuring mean acid-precipitable [3H]thymidine incorporation (10µCi/ml in growth medium) for five sequential 1-h incubations, in duplicate, for each cell cycle fraction identified in Fig. 3. Note that each cell fraction reaches S phase at sequentially later times after return to culture.](images/v1_pa_s07_c31_f04.jpg) |
| FIGURE 4 Analysis of synchrony of entry into DNA synthesis in centrifugal elutriation fractions of HeLa S3 cells. HeLa cells were separated into synchronous fractions by centrifugal elutriation, and kinetics of entry into DNA synthesis phase (S phase) was determined by measuring mean acid-precipitable [3H]thymidine incorporation (10µCi/ml in growth medium) for five sequential 1-h incubations, in duplicate, for each cell cycle fraction identified in Fig. 3. Note that each cell fraction reaches S phase at sequentially later times after return to culture. |
- Analyze cell cycle fractions by flow cytometric analysis (Fig. 3). Wash approximately 5 × 105 cells by centrifugation and resuspend in ice-cold Hanks' balanced salt solution. Fix cells by addition of an equal volume of ice-cold 4% paraformaldehyde. After a 24- h incubation at 4°C collect the cells by centrifugation and resuspend in 2 ml of ice-cold PBS. Alternatively, fix cells by slow dropwise addition of 3 volumes of 70% ethanol (-20°C while applying continuous gentle agitation with a vortex mixer. Allow cells to fix for at least 1 h at 4°C and then wash as described earlier. Approximately 30min prior to flow cytometric analysis, add 3ml of staining solution to each aliquot of 300µl of cell suspension and incubate at room temperature. Analyze fluorescence on a flow cytometer (Elite Flow Cytometer, Coulter Electronics).
- Analyze acid precipitable [3H]thymidine incorporation by synchronous cell populations by pulse labeling each separated fraction of cells with 10 µCi/ml [3H]thymidine in complete growth medium (Fig. 4) for 1 h at hourly intervals after return of the cells to culture (Wu et al., 1993; Bird et al., 1988). Place cells in 96-well plates (2 × 104/well) in 100µl growth medium and incubate under normal growth conditions. Add 1 µCi [3H]thymidine to each well at the appropriate time and incubate for 1 h at 37°C. Wash each cell fraction twice with Hanks' balanced salt solution, drain fluid, lyse in 100µl TES, load 100btl of lysate onto a 2.4-cm filter paper circle (Whatman 540, Cat. No. 1540-324) labeled with a No. 2 pencil, and allow to air dry. Precipitate samples with excess solutions of 200ml for up to 50 filters for 20min (do not exceed this time or the filters may be damaged): 20% TCA, 10% TCA, absolute ethanol, ether, absolute ethanol. Then, air dry and determine the radioactivity in each sample by liquid scintillation counting in a 5-ml ScintiSafe Econ 1 scintillation fluor (Fisher, Cat. No. SX20-5) as described previously (Wu et al., 1993; Bird et al., 1988).
A. Theoretical Basis of Separation
Cells are separated in the centrifugal elutriator by a process that was originally called counterstream centrifugation. This process is based on the opposition of two forces: media flow and angular acceleration due to an applied centripetal force. All of the cells in the suspension to be separated by centrifugal elutriation are pumped under gentle pressure into the separation chamber, entering at the extreme outside end of the chamber where the radius of rotation is maximal (rmax) to begin accumulating in the chamber (Fig. 1). As the cells fill the chamber, they change direction (as the loading tube turns) from flowing out to the perimeter of the rotor to flowing toward the center of the rotor. As they do, the particles enter a chamber that is designed with a conical profile. As the particles enter the chamber, the cross-sectional area increases rapidly, dramatically decreasing inward movement of the cells under a constant flow rate. Thus, the cells begin to decelerate as the cross-sectional area of the chamber increases. At some point, determined by cell size, media flow rate and viscosity, and centripetal force, the cells cease to move inward and become suspended in the chamber at a position where cell movement inward due to flow rate is balanced by apparent centrifugal force outward. At this point, medium is flowing by the cells but inward movement of the cells is offset by the apparent outward centrifugal force applied due to the angular momentum of the rotor. The population of cells has thus reached equilibrium and their position remains unchanged, although the cells tend to sort themselves under this combination of opposing forces with the smallest particles sorting closest to the center of the rotor.
The actual elutriation characteristics depend on the shape and size of the cells in the population. For trypsinized cells, whose specific gravity is not much greater than the buffer in which they are suspended, shape generally approaches spherical unless cell processes or applied forces alter this. Thus, separation is usually the product of apparent cell diameter. Centrifugal elutriation has been shown to be unusually subtle in its ability to discriminate subfractions based on very small differences in cell size. For example, in separation of cells in different phases of the cell cycle, cells approximately double in volume as they pass through the cell cycle from G1 phase to mitosis (Mitchison, 1971). This translates into an increase of only about 26% in the radius of an average premitotic cell compared to an average freshly divided G1 phase cell. This relationship can be demonstrated by the following equations where V is cell volume and r is cell radius, assuming a spherical shape. The G1 phase cell is represented by a volume of VG1 and an apparent radius of rG1, the premitotic cell is represented by a volume of VM and an apparent radius of rM, and the assumption is made that cell volume exactly doubles on average during the intervening period.
Thus
| 4/3 π (rG1)3 = VG1 | |
| 1 (volume of a spherical G1 phase cell) | (1) |
| 4/3 π (rG1)3 = VM | |
| 2 (volume of a spherical premitotic cell) | (2) |
| 2 VG1 = VM | |
| 3 (volume doubles during the cell cycle) | (3) |
| Substituting Eqs. (1) and (2) into Eq. (3) for each volume and then simplifying, | |
| 2[4/3 π (rG1)3] = [4/3 π (rM)3] | |
| 2 (rG1)3 = (rM)3 | |
Taking the cube root of each side, | |
| 1.26 rG1 = rM | |
The theoretical basis on which centrifugal elutriation is capable of separating cells, by balancing changes in medium flow balanced against angular momentum, has revealed that sequential fractions were frequently differentiated by as little as a 3% difference in cell diameter. In most cases, apart from the expense of purchase of the elutriation centrifuge, the only compromise consists of a somewhat lower level of purity in some samples, although purities approaching 100% have been reported. To balance this drawback, great improvements in yield, speed, and gentleness of handling of the cells have been achieved. Due to these obvious benefits, centrifugal elutriation has been applied to a wide variety of cells and cell types to successfully affect separation based on cell size.
V. PITFALLS
- In the time since we acquired our centrifugal elutriator, Beckman Instruments has released newer models designed to separate, among other qualities, larger numbers of cells. While the principles governing separation remain the same, details of procedures necessary in setting up the rotor and the seals may differ, depending on the model, and thus the manufacturer's directions should be adhered to carefully to ensure correct assembly and operation.
- It is critical that separations be attempted only with single cell populations. Steps should be taken to ensure complete dissociation of cells liberated from tissues or culture vessels. When in doubt, dispersion of samples should be monitored microscopically.
- Only Beckman neutral pH rotor detergent should be used to clean the rotor and separation chamber. It is particularly important to ensure that the sample tube at the outer edge of the separation chamber is soaked in detergent overnight to remove any cell debris that has accumulated as this will affect flow rate greatly as well as the fluid dynamics of sample loading in the chamber. This aperture is too small to be cleaned with a tool, and no instrument should be applied that could scratch the interior surface of the chamber. The operator must be extremely careful with the separation chamber, as overtightening of the screws or scratches to the surface will damage the chamber, affecting performance significantly.
- If alterations to the growth medium or elutriation medium are contemplated, the new medium must be tested to empirically determine at what flow rate cell cycle fractions elute. Even very small changes in media formulation (e.g., as little as 0.5% change in DHS concentration) will change the fluid dynamics of the elutriation system dramatically. Changes in cell-loading density and temperature can also create detectable effects on elutriation profiles. A simple pilot experiment is usually sufficient to determine what effects such alterations have on elution flow rate if it is followed by flow cytometric analysis.
- All O rings and gaskets should be inspected before each run to ensure that they are in good condition. All worn seals should be replaced.
- Failure to secure the wires and tubing connecting the rotor and strobe light to the exterior of the centrifuge will result in them becoming wrapped around the rotor, resulting in serious damage to the equipment.
- We have replaced the Oakridge-style sample application tube supplied by the manufacturer with a straight glass test tube (13 × 100mm) with the same size aperture at the top as the tube supplied. This eliminates the shoulder at the top of the Oakridge tube, which can trap cells, resulting in a continuous residual loading of cells throughout the elutriation procedure. The tube should be siliconized to within approximately 2cm from the top with Gel Slick (FMC Bio- Products). Gel Slick should not be allowed to contact the glass surface above this point as the stopper will no longer hold securely under pressure.
- Rotor speed should be monitored frequently during the run as fluctuations of only a small amount can affect purity of the samples greatly. It is particularly important to check rotor speed each time that the refrigeration system in the centrifuge cycles on.
- If sterilization of the assembly is required, be sure that the centrifuge is turned off while the ethanol is in the system. Failure to observe this safety measure could result in a fire hazard. Alternatively, the system can be sterilized with 6% hydrogen peroxide while the centrifuge is running (Conkie, 1985).
Acknowledgments
The author thanks Dr. Gin Wu, Dr. Suresh Pai, and Dr. Shiawhwa Su for consultation on this protocol and Patricia DeInnocentes and Randy Young-White for valuable technical support. The author also thanks Dr. Lauren G. Wolfe for critical reading of the manuscript.
Beckman Instruments (1990). Centrifugal Elutriation of Living Cells: An Annotated Bibliography. In Applications Data, Number DS- 534, pp. 1-41, Beckman Instruments Inc., Palo Alto, CA.
Bialkowski, K., and Kasprzak, K. S. (2000). Activity of the antimutagenic enzyme 8-oxo-2'-deoxyguanosine 5'-triphosphate pyrophosphohydrolase (8-oxo-dGTPase) in cultured chinese hamster ovary cells: Effects of cell cycle, proliferation rate, and population density. Free Radic. Biol. Med. 28, 337-344.
Bird, R. C. (1996a). Cell separation by centrifugal elutriation. In "Cell Biology: A Laboratory Handbook" (J. E. Celis, ed.), 2nd Ed., pp. 205-208. Academic Press, New York.
Bird, R. C. (1996b). Synchronous populations of cells in specific phases of the cell cycle prepared by centrifugal elutriation. In "Cell Biology: A Laboratory Handbook" (J. E. Celis, ed.), 2nd Ed., pp. 209-217. Academic Press, New York.
Bird, R. C., Bartol, E E, Daron, H., Stringfellow, D. A., and Ridell, G. M. (1988). Mitogenic activity in ovine uterine fluids: Characterization of a growth factor which specifically stimulates myoblast proliferation. Biochem. Biophys. Res. Commun. 156, 108-115.
Bludau, M., Kopun, M., and Werner, D. (1986). Cell cycle-dependent expression of nuclear matrix proteins of Ehrlich ascites cells studied by in vitro translation. Exp. Cell Res. 165, 269-282.
Braunstein, J. D., Schulze, D., DelGiudice, T., Furst, A., and Schildkraut, C. L. (1982). The temporal order of replication of murine immunoglobulin heavy chain constant region sequences corresponds to their linear order in the genome. Nucleic. Acids Res. 10, 6887-6902.
Brown, E. H., and Schildkraut, C. L. (1979). Perturbation of growth and differentiation of Friend murine erythroleukemia cells by 5-bromodeoxyuridine incorporation in early S phase. J. Cell. Physiol. 99, 261-277.
Conkie, D. (1985). Separation of viable cells by centrifugal elutriation. In "'Animal Cell Culture: A Practical Approach" (R. I. Freshney, ed.), pp. 113-124. IRL Press, Oxford.
Dagher, R., Long, L. M., Read, E. J., Leitman, S. E, Carter, C. S., Tsokos, M., Goletz, T. J., Avila, N., Berzofsky, J. A., Helman, L. J., and Mackall, C. L. (2002). Pilot trial of tumor-specific peptide vaccination and continuous infusion interleukin-2 in patients with recurrent Ewing sarcoma and alveolar rhabdomyosarcoma: An inter-institute NIH study. Med. Pediatr. Oncol. 38, 158-164.
Datta, N. S., and Long, M. W. (2002). Modulation of MDM2/p53 and cyclin-activating kinase during the megakaryocyte differentiation of human erythroleukemia cells. Exp. Hematol. 2, 158-165. Davis, P. K., Ho, A., and Dowdy, S. E (2001). Biological methods for cell-cycle synchronization of mammalian cells. Biotechniques 30, 1322-1331.
Diamond, R. A. (1991) Separation and enrichment of cell populations by centrifugal elutriation. Methods 2, 173-182. Hann, S. R., Thompson, C. B., and Eisenman, R. N. (1985) c-myc oncogene protein synthesis is independent of the cell cycle in human and avian cells. Nature 314, 366-369.
Hengstschlager, M., Holzl, G., and Hengstschlager-Ottnad, E. (1999). Different regulation of c-Myc- and E2F-l-induced apoptosis during the ongoing cell cycle. Oncogene 18, 843-848.
Houser, S., Koshlatyi, S., Lu, T., Gopen, T., and Bargonetti, J. (2001). Camptothecin and zeocin can increase p53 levels during all cell cycle stages. Biochem. Biophys. Res. Commun. 289, 998-1009.
Iqbal, M. A., Plumb, M., Stein, J., Stein, G., and Schildkraut, C. L. (1984). Coordinate replication of members of the multigene family of core and H1 human histone genes. Proc. Natl. Acad. Sci. USA 81, 7723-7727.
Karas, M., Zaks, T. Z., Liu, J. L., and LeRoith, D. (1999). T cell receptor- induced activation and apoptosis in cycling human T cells occur throughout the cell cycle. Mol. Biol. Cell 10, 4441-4450.
Lindahl, P. E. (1948). Principle of counterstreaming centrifuge for the separation of particles of different sizes. Nature 161, 648-649.
Lindahl, P. E. (1986). On counterstreaming centrifugation in the separation of cells and cell fragments. Biochim. Biophys. Acta 21, 411-415.
Lindberg, C. A. (1932). A method for washing corpuscles in suspension. Science 75, 415-416.
Merrill, G. E (1998). Cell synchronization. Methods Cell Biol. 57, 229-249.
Mitchison, J. M. (1971). "The Biology of the Cell Cycle." Cambridge Univ. Press, Cambridge.
Pai, S. R., and Bird, R. C. (1992). Growth of HeLa $3 cells cotransfected with plasmids containing a c-fos gene under the control of the SV40 promoter complex, pRSVcat, and G418 resistance. Biochem. Cell Biol. 70, 316-323.
Pai, S. R., and Bird, R. C. (1994). Overexpression of c-fos induces expression of the retinoblastoma tumor suppressor gene in transfected cells. Anticancer Res. 14, 2501-2508.
Rehak, M., Csuka, I., Szepessy, E., and Banfalvi. G. (2000). Subphases of DNA replication in Drosophila cells. DNA Cell Biol. 19, 607-612.
Sambrook, J., Fritsch, E. R, and Maniatis, T. (1989). "Molecular Cloning: A Laboratory Manual" (J. Sambrook, E. E Fritsch, and T. Maniatis, eds.), 2nd Ed., p B.17. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
Syljuasen, R. G., and McBride, W. H. (1999). Radiation-induced apoptosis and cell cycle progression in Jurkat T cells. Radiat. Res. 152, 328-331.
Van Leeuwen-Stok, E. A., Jonkhoff, A. R., Visser-Platier, A. W., Drager, L. M., Teule, G. J., Huijgens, P. C., and Schuurhuis, G. J. (1998). Cell cycle dependency of 67gallium uptake and cytotoxicity in human cell lines of hematological malignancies. Leuk. Lymphoma 31, 533-544.
Wong, E. C., Lee, S. M., Hines, K., Lee, J., Carter, C. S., Kopp, W., Bender, J., and Read, E. J. (2002). Development of a closed-system process for clinical-scale generation of DCs: Evaluation of two monocyte-enrichment methods and two culture containers. Cytotherapy 4, 65-76.
Wong, E. C., Maher, V. E., Hines, K., Lee, J., Carter, C. S., Goletz, T., Kopp, W., Mackall, C. L., Berzofsky, J., and Read, E. J. (2001). Development of a clinical-scale method for generation of dendritic cells from PBMC for use in cancer immunotherapy. Cytotherapy 3, 19-29.
Wu, G., Su, S., Kung, T.-Y. T., and Bird, R.C. (1993). Molecular cloning of Gl-phase mRNAs from a subtractive Gl-phase cDNA library. Biochem. Cell Biol. 71, 372-380.
Support our developers




