Dihydrofolate Reductase
Dihydrofolate reductase (DHFR) catalyzes the reduction of 7,8-dihydrofolate (H2F) by nicotinamide adenine dinucleotide phosphate (reduced form) (NADPH) to form 5,6,7,8-tetrahydrofolate (H4F), a key step in furnishing the parental cofactor needed for de novo pyrimidine and purine biosynthesis. The enzyme has been the target of antitumor and antimicrobial drugs. A complete kinetic scheme (Fig. 6) obtained primarily through transient kinetics has been described for the enzyme from Escherichia coli as well as other sources and provides a second case study as to how to define the catalytic process.Measurement of the rates of binding and dissociation of substrate and cofactors provided valuable insights into the identity of rate-limiting kinetic steps in the scheme shown in Fig. 6. Two procedures were used. In the first, direct observation of changes in the intrinsic enzyme or NADPH fluorescence upon ligand binding showed that the addition of ligand was biphasic in accord with the existence of two conformers, of which only one bound the ligand:
| DHFR1 | k2 | DHFR2 | k1 | DHFR2 | . L. | |||
| + L | ||||||||
| k−2 | k−1 |
| DHFR | . L. | k−1 | DHFR | kT[T] | DHFR | . T. | |||
| + L1 | |||||||||
| k1 | k−T |
procedure identified a preferred pathway for dissociation of the product H4F as the rate-limiting step in the steady-state cycle. The assistance of the cofactor NADPH in promoting product dissociation is an unusual feature, though not limited to DHFR, and follows the rapid loss of NADP+.
Events around the chemical step of reduction/oxidation were monitored by directly observing the conversion of NADPH to NADP+. The kinetics are again biphasic owing to the rapidity of the hydride transfer process; that the rapid phase is associated with the chemical step is verified by the observation of a kinetic deuterium isotope effect of 3 when the transferring hydrogen of the NADPH is replaced with deuterium. This step shows a pH dependence with a pKa of 6.5 that implicates the Asp 125 (27 in E. coli) in the proton transfer events required to complete the reduction. Measurement of this step in the reverse direction (i.e., for DHFR · NADP+ ·H4F) coupled with determination of the overall equilibrium constant permitted construction of Fig. 6.
The kinetic scheme served as the basis for the explanation of the contribution of various elements of the protein to its function. Site-specific mutagenesis is a technique in which one or more amino acids are replaced by other amino acids through alteration of the gene encoding the enzyme. For the mutant proteins, the same kinetic scheme was reconstructed to calculate the free energy differences arising from changes in the kinetic steps caused by the mutations. Replacing the hydrophobic residues such as Phe30 and Leu54 (Fig. 7) singly or pairwise with other amino acids revealed that the cumulative effect of two mutations was generally nonadditive in terms of the free energy associated with individual steps in Fig. 6, consistent with long-range interactions across the enzyme active site mediated by bound substrate and cofactor. The nonadditivity differed for each step in Fig. 6, which implicated differing conformations of the protein as arising throughout the catalytic cycle.
Of particular interest was the discovery that changes in the amino acid sequence at loci outside the active site also strongly influence (by a factor of >102) the rate of the chemical step. In combination with dynamic NMR measurements and molecular mechanics calculations, this observation has been attributed to the importance for catalysis of long-range motions that occur across the entire DHFR protein molecule. The protein fold through its complex vibrational modes apparently may couple some set of motions to a promotional vibration that fosters passage of the reactive ternary complex over the activation barrier.
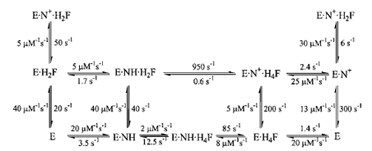 |
| Figure 6 The kinetic scheme for conversion of H2F to H4F by DHFR, including the rate constants for each step at 25°C. In this scheme, NH represents NADPH and N+ represents NADP+. |
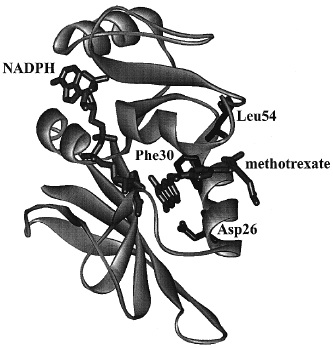 |
| Figure 7 Crystal structure of DHFR from Lactobacillus casei with methotrexate (a strong inhibitor) and NADPH bound. Amino acid residues discussed in the text are labeled. |
Support our developers




