Taxonomic Characters and Phylogenetic Reconstruction
Taxonomic
Characters and
Phylogenetic
Recostruction
A major goal of systematics is to reconstruct the evolutionary tree or phylogeny that relates all extant and extinct species. This task is accomplished by studying organismal features, formally called characters, that vary among species. A character is any feature that the taxonomist uses to study variation within and among species. We find potentially useful taxonomic characters in morphological, chromosomal, and molecular features. Taxonomists find characters by observing patterns of similarity among organisms. If two organisms possess similar features, they may have inherited these features from a common ancestor. Character similarity that results from common ancestry is called homology (see Organic Evolution). Similarity does not always reflect common ancestry, however. Independent evolutionary origin of similar features on different lineages produces patterns of similarity among organisms that do not reflect common descent; this occurrence complicates the work of taxonomists. Character similarity that misrepresents common descent is called nonhomologous similarity or homoplasy.
Using Character Variation to Reconstruct Phylogeny
To reconstruct the phylogeny of a group using characters that vary among its members, the first step is to determine which variant form of each character was present in the common ancestor of the entire group. This character state is called ancestral for the group as a whole. We presume that all other variant forms of the character arose later within the group, and these are called evolutionarily derived character states. The polarity of a character refers to the ancestral/descendant relationships among its different states. For example, if we consider as a character the dentition of amniotic vertebrates (reptiles, birds, and mammals), presence versus absence of teeth in the jaws constitute two different character states. Teeth are absent from birds but present in the other amniotes. To evaluate the polarity of this character, we must determine which character state, presence or absence of teeth, characterized the most recent common ancestor of amniotes and which state was derived subsequently within the amniotes.
The method that we use to examine the polarity of a variable character is called outgroup comparison. We consult an additional group of organisms, called an outgroup, that is phylogenetically close but not within the group being studied. We infer that any character state found both within the group being studied and in the outgroup is ancestral for the study group. The amphibians and different groups of bony fishes constitute appropriate outgroups to the amniotes for polarizing variation in the dentition of amniotes. Teeth are usually present in amphibians and bony fishes; therefore, we infer that presence of teeth is ancestral for amniotes and absence of teeth is derived. The polarity of this character indicates that teeth were lost in the ancestral lineage of all modern birds. Polarity of characters is evaluated most effectively when several different outgroups are used. All character states found in the study group that are absent from appropriate outgroups are considered derived.
The organisms or species that share derived character states form subsets within the group called clades (Gr. klados, branch). A derived character shared by the members of a clade is formally called a synapomorphy (Gr. synapsis, joining together, + morphe, form) of that clade. Taxonomists use synapomorphies as evidence of homology to infer that a particular group of organisms forms a clade. Within the amniotes, absence of teeth and presence of feathers are synapomorphies that identify the birds as a clade. A clade corresponds to a unit of evolutionary common descent; it includes all descendants of a particular ancestral lineage. The pattern formed by the derived states of all characters within our study group will take the form of a nested hierarchy of clades within clades. The goal is to identify all of the different clades nested within the study group, which would give a complete account of the patterns of common descent among the species in the group.
Character states ancestral for a taxon are often called plesiomorphic for that taxon and the sharing of ancestral states among organisms is termed symplesiomorphy. Unlike synapomorphies, however, symplesiomorphies do not provide useful information on nesting of clades within clades. In the example given above, we found that presence of teeth in jaws was plesiomorphic for amniotes. If we grouped together mammalian and reptilian groups, which possess teeth, to the exclusion of birds, we would not obtain a valid clade. Birds also descend from all common ancestors of reptiles and mammals and must be included within any clade that includes all reptiles and mammals. Errors in determining polarity of characters therefore clearly can produce errors in inference of phylogeny. It is important to note, however, that character states that are plesiomorphic at one taxonomic level can be synapomorphies at a more inclusive level. For example, the presence of jaws bearing teeth is a synapomorphy of gnathostome vertebrates, a group that includes the amniotes plus amphibians, bony fishes, and cartilaginous fishes, although teeth have been lost in birds and some other gnathostomes. The goal of phylogenetic analysis therefore can be restated as one of finding the appropriate taxonomic level at which any given character state is a synapomorphy. The character state is then used at that level to identify a clade.
The nested hierarchy of clades is presented as a branching diagram called a cladogram (Figure 10-2; see also Figure 6-15). Taxonomists often make a technical distinction between a cladogram and a phylogenetic tree. The branches of a cladogram are only a formal device for indicating the nested hierarchy of clades within clades. The cladogram is not strictly equivalent to a phylogenetic tree, whose branches represent real lineages that occurred in the evolutionary past. To obtain a phylogenetic tree, we must add to the cladogram important additional information concerning ancestors, the durations of evolutionary lineages, or the amounts of evolutionary change that occurred on the lineages. Because the structure of a cladogram is congruent with that of the corresponding phylogenetic tree, however, the cladogram is often used as a first approximation of the phylogenetic tree.
Sources of Phylogenetic Information
We find characters used to construct cladograms in comparative morphology (including embryology), comparative cytology, and comparative biochemistry. Comparative morphology examines the varying shapes and sizes of organismal structures, including their developmental origins. As we will see in later sections, the variable structures of skull bones, limb bones, and integument (scales, hair, feathers) are particularly important for reconstructing the phylogeny of vertebrates. Comparative morphology uses specimens obtained from both living organisms and fossilized remains. Comparative biochemistry uses sequences of amino acids in proteins and the sequences of nucleotides in nucleic acids (see Principles of Genetics:A Review) to identify variable characters for constructing a cladogram (Figure 10-3). Direct sequencing of DNA is regularly applied to phylogenetic studies; however, comparisons of protein sequences are usually indirect, involving immunological or allozymic (see Figure 6-30) methods, or inference from DNA sequences of protein-coding genes. Recent studies have shown that comparative biochemistry can be applied to some fossils in addition to living organisms. Comparative cytology uses variation in the numbers, shapes, and sizes of chromosomes and their parts (Cells as Units of Life) to obtain variable characters for constructing cladograms. Comparative cytology is used almost exclusively on living rather than fossilized organisms.
To add an evolutionary timescale necessary for producing a phylogenetic tree, we must consult the fossil record. We can look for the earliest appearance in fossils of derived morphological characters to estimate the ages of clades distinguished by those characters. The age of a fossil showing the derived characters of a particular clade is determined by radioactive dating. An example of a phylogenetic tree constructed using these methods is Figure 21-6.
We can use comparative biochemical data to estimate the ages of different lineages on a phylogenetic tree. Some protein and DNA sequences undergo approximately linear rates of divergence through evolutionary time. The age of the most recent common ancestor of two species is therefore proportional to the differences measured between their proteins and DNA sequences. We calibrate evolution of proteins and DNA sequences by measuring their divergence between species whose most recent common ancestor has been dated using fossils. We then use the molecular evolutionary calibration to estimate ages of other branches on the phylogenetic tree.
A major goal of systematics is to reconstruct the evolutionary tree or phylogeny that relates all extant and extinct species. This task is accomplished by studying organismal features, formally called characters, that vary among species. A character is any feature that the taxonomist uses to study variation within and among species. We find potentially useful taxonomic characters in morphological, chromosomal, and molecular features. Taxonomists find characters by observing patterns of similarity among organisms. If two organisms possess similar features, they may have inherited these features from a common ancestor. Character similarity that results from common ancestry is called homology (see Organic Evolution). Similarity does not always reflect common ancestry, however. Independent evolutionary origin of similar features on different lineages produces patterns of similarity among organisms that do not reflect common descent; this occurrence complicates the work of taxonomists. Character similarity that misrepresents common descent is called nonhomologous similarity or homoplasy.
Using Character Variation to Reconstruct Phylogeny
To reconstruct the phylogeny of a group using characters that vary among its members, the first step is to determine which variant form of each character was present in the common ancestor of the entire group. This character state is called ancestral for the group as a whole. We presume that all other variant forms of the character arose later within the group, and these are called evolutionarily derived character states. The polarity of a character refers to the ancestral/descendant relationships among its different states. For example, if we consider as a character the dentition of amniotic vertebrates (reptiles, birds, and mammals), presence versus absence of teeth in the jaws constitute two different character states. Teeth are absent from birds but present in the other amniotes. To evaluate the polarity of this character, we must determine which character state, presence or absence of teeth, characterized the most recent common ancestor of amniotes and which state was derived subsequently within the amniotes.
The method that we use to examine the polarity of a variable character is called outgroup comparison. We consult an additional group of organisms, called an outgroup, that is phylogenetically close but not within the group being studied. We infer that any character state found both within the group being studied and in the outgroup is ancestral for the study group. The amphibians and different groups of bony fishes constitute appropriate outgroups to the amniotes for polarizing variation in the dentition of amniotes. Teeth are usually present in amphibians and bony fishes; therefore, we infer that presence of teeth is ancestral for amniotes and absence of teeth is derived. The polarity of this character indicates that teeth were lost in the ancestral lineage of all modern birds. Polarity of characters is evaluated most effectively when several different outgroups are used. All character states found in the study group that are absent from appropriate outgroups are considered derived.
The organisms or species that share derived character states form subsets within the group called clades (Gr. klados, branch). A derived character shared by the members of a clade is formally called a synapomorphy (Gr. synapsis, joining together, + morphe, form) of that clade. Taxonomists use synapomorphies as evidence of homology to infer that a particular group of organisms forms a clade. Within the amniotes, absence of teeth and presence of feathers are synapomorphies that identify the birds as a clade. A clade corresponds to a unit of evolutionary common descent; it includes all descendants of a particular ancestral lineage. The pattern formed by the derived states of all characters within our study group will take the form of a nested hierarchy of clades within clades. The goal is to identify all of the different clades nested within the study group, which would give a complete account of the patterns of common descent among the species in the group.
Character states ancestral for a taxon are often called plesiomorphic for that taxon and the sharing of ancestral states among organisms is termed symplesiomorphy. Unlike synapomorphies, however, symplesiomorphies do not provide useful information on nesting of clades within clades. In the example given above, we found that presence of teeth in jaws was plesiomorphic for amniotes. If we grouped together mammalian and reptilian groups, which possess teeth, to the exclusion of birds, we would not obtain a valid clade. Birds also descend from all common ancestors of reptiles and mammals and must be included within any clade that includes all reptiles and mammals. Errors in determining polarity of characters therefore clearly can produce errors in inference of phylogeny. It is important to note, however, that character states that are plesiomorphic at one taxonomic level can be synapomorphies at a more inclusive level. For example, the presence of jaws bearing teeth is a synapomorphy of gnathostome vertebrates, a group that includes the amniotes plus amphibians, bony fishes, and cartilaginous fishes, although teeth have been lost in birds and some other gnathostomes. The goal of phylogenetic analysis therefore can be restated as one of finding the appropriate taxonomic level at which any given character state is a synapomorphy. The character state is then used at that level to identify a clade.
The nested hierarchy of clades is presented as a branching diagram called a cladogram (Figure 10-2; see also Figure 6-15). Taxonomists often make a technical distinction between a cladogram and a phylogenetic tree. The branches of a cladogram are only a formal device for indicating the nested hierarchy of clades within clades. The cladogram is not strictly equivalent to a phylogenetic tree, whose branches represent real lineages that occurred in the evolutionary past. To obtain a phylogenetic tree, we must add to the cladogram important additional information concerning ancestors, the durations of evolutionary lineages, or the amounts of evolutionary change that occurred on the lineages. Because the structure of a cladogram is congruent with that of the corresponding phylogenetic tree, however, the cladogram is often used as a first approximation of the phylogenetic tree.
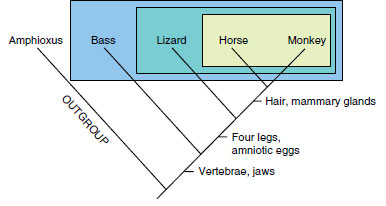 |
| Figure 10-2 The cladogram as a nested hierarchy of taxa. Amphioxus is the outgroup, and the study group comprises four vertebrates (bass, lizard, horse, and monkey). Four characters that vary among vertebrates are used to generate a simple cladogram: presence versus absence of four legs, amniotic eggs, hair, and mammary glands. For all four characters, absence is the ancestral state in vertebrates because this is the condition found in the outgroup, Amphioxus; for each character, presence is the derived state in vertebrates. Because they share presence of four legs and amniotic eggs as synapomorphies, the lizard, horse, and monkey form a clade relative to the bass. This clade is subdivided further by two synapomorphies (presence of hair and mammary glands) that unite the horse and monkey relative to the lizard. We know from comparisons involving even more distantly related animals that presence of vertebrae and jaws constitute synapomorphies of vertebrates and that Amphioxus, which lacks these features, falls outside the vertebrate clade. |
Sources of Phylogenetic Information
We find characters used to construct cladograms in comparative morphology (including embryology), comparative cytology, and comparative biochemistry. Comparative morphology examines the varying shapes and sizes of organismal structures, including their developmental origins. As we will see in later sections, the variable structures of skull bones, limb bones, and integument (scales, hair, feathers) are particularly important for reconstructing the phylogeny of vertebrates. Comparative morphology uses specimens obtained from both living organisms and fossilized remains. Comparative biochemistry uses sequences of amino acids in proteins and the sequences of nucleotides in nucleic acids (see Principles of Genetics:A Review) to identify variable characters for constructing a cladogram (Figure 10-3). Direct sequencing of DNA is regularly applied to phylogenetic studies; however, comparisons of protein sequences are usually indirect, involving immunological or allozymic (see Figure 6-30) methods, or inference from DNA sequences of protein-coding genes. Recent studies have shown that comparative biochemistry can be applied to some fossils in addition to living organisms. Comparative cytology uses variation in the numbers, shapes, and sizes of chromosomes and their parts (Cells as Units of Life) to obtain variable characters for constructing cladograms. Comparative cytology is used almost exclusively on living rather than fossilized organisms.
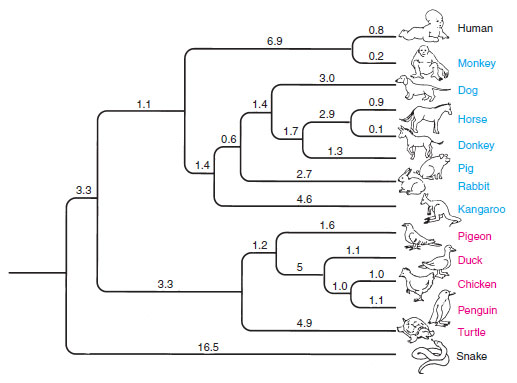 |
| Figure 10-3 A phylogenetic tree of representative amniotes based on inferred base substitutions in the gene that encodes the respiratory protein, cytochrome c. Numbers on the branches are the estimated numbers of mutational changes that occurred in this gene along the different evolutionary lineages. |
To add an evolutionary timescale necessary for producing a phylogenetic tree, we must consult the fossil record. We can look for the earliest appearance in fossils of derived morphological characters to estimate the ages of clades distinguished by those characters. The age of a fossil showing the derived characters of a particular clade is determined by radioactive dating. An example of a phylogenetic tree constructed using these methods is Figure 21-6.
We can use comparative biochemical data to estimate the ages of different lineages on a phylogenetic tree. Some protein and DNA sequences undergo approximately linear rates of divergence through evolutionary time. The age of the most recent common ancestor of two species is therefore proportional to the differences measured between their proteins and DNA sequences. We calibrate evolution of proteins and DNA sequences by measuring their divergence between species whose most recent common ancestor has been dated using fossils. We then use the molecular evolutionary calibration to estimate ages of other branches on the phylogenetic tree.
Support our developers




