Genomic DNA Microarray for Comparative Genomic Hybridization
DNA copy number aberrations are characteristic of many human cancers as well as developmental abnormalities. Array comparative genomic hybridization (CGH) can be used to quantitatively and precisely detect these DNA copy number aberrations (Solinas- Toldo et al., 1997; Pinkel et al., 1997). Bacterial artificial chromosome (BAC) clones have proven to yield sufficient signal intensity after hybridization to detect lowlevel DNA copy number aberrations as well as high-level amplifications (Snijders et al., 2001). Also, when BAC clones are used that are mapped onto the sequence of the human genome, one can link aberrations directly onto the sequence.
However, because BAC clones are single copy vectors, their yield from a bacterial culture is relatively low, making it tedious to produce sufficient quantities of BAC DNA necessary to quantitatively detect DNA copy number aberrations. In addition, reliably depositing high molecular weight BAC DNA can be challenging due to the viscosity of the printing solution caused by the high concentration of the high molecular weight BAC DNA.
This article describes a polymerase chain reaction (PCR)-based method (Klein et al., 1999) for producing large quantities of BAC DNA, which aims at maximizing the representation of each BAC (Snijders et al., 2001). An overview of the procedure is shown in Fig. 1. In short, BAC DNA is cut with a frequently cutting restriction enzyme. Primers are ligated to the overhanging ends created by cleavage of the BAC DNA. A subsequent PCR is carried out to amplify the BAC DNA. Using only a fraction of the first PCR, a second PCR is performed, and this DNA product is made into spotting solution. This article also describes the DNA labeling and hybridization protocols used to obtain highly reproducible and quantitative data.
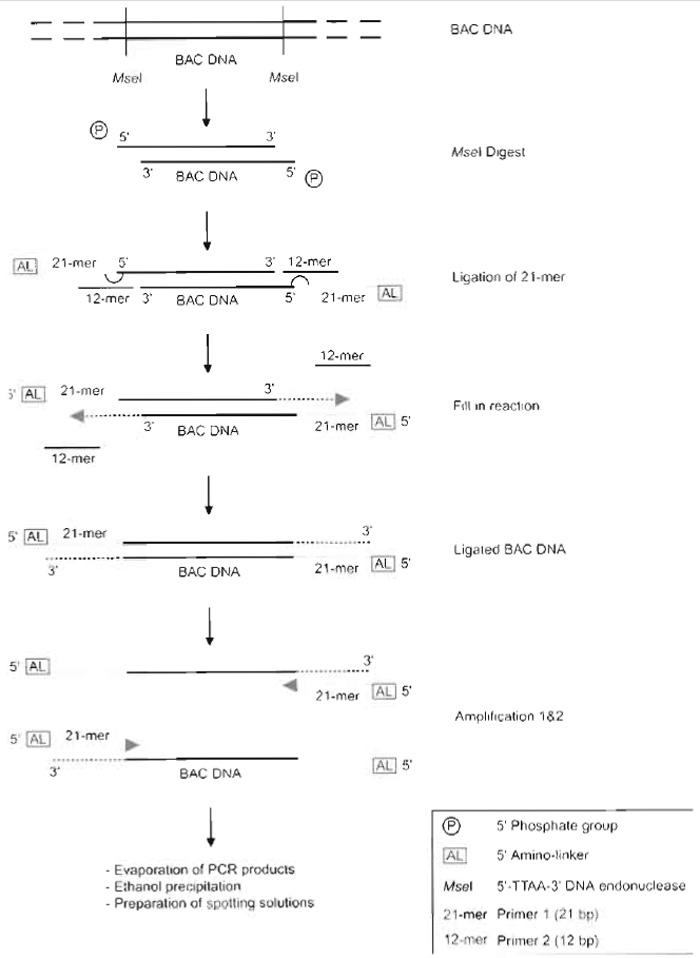 |
| FIGURE 1 Overview of the ligation-mediated PCR procedure. BAC DNA is digested with the MseI restriction enzyme, leaving a 5'-phosphorylated TA overhang. Primers are ligated to these overhangs. The 12-bp primer guides the amino linker containing the 21-bp primer to the overhang where the 21-bp primer is ligated by T4 DNA ligase. Because of the absence of a phosphate group at the 5' end of the 12-bp primer, it will not get ligated to the BAC DNA. Primer 2 is melted off, after which DNA polymerase fills in the now singlestranded DNA overhang (as indicated with a dashed arrow). The now double-stranded, BAC DNA fragments are PCR amplified using a high-fidelity DNA polymerase; excess primer 2 from the ligation reaction will prime the reaction. A fraction of this ligation-mediated PCR is amplified in a second PCR reaction, which will be made into spotting solution. |
A. Preparation of BAC DNA Spotting Solutions
10x One-Phor-All Buffer Plus [100 mM Tris-acetate (pH 7.5), 100mM magnesium acetate and 500mM potassium acetate] is from Amersham Pharmacia Biotech Inc. (Cat. No. 27-0901-02) and is stable at 4°C
MseI restriction enzyme (50U/µl) is from New England Biolabs (Cat. No. R0525M). Store at -20°C. Store on ice when in use return to -20°C as soon as possible.
20-500ng of BAC DNA with minimal contamination from host DNA. A Qiagen plasmid minikit (Cat. No. 12125) can be used to purify BAC DNA by following a modification of the manufacturer's protocol (see Comment 1)
0.2-ml polypropylene PCR eight-tube strips with separate eight-cap strips (Cat. No. 1402-2700) and 96- well plate seal (Cat. No. 2920-0000) are from USA Scientific
Ultrapure agarose (Cat. No. 15510-027), ATP (10mM) (Cat. No. 18330-019), T4 DNA ligase (5U/µl) (Cat. No. 15524-041), dATP, dCTP, dGTP, and dTTP (100mM each) (Cat. No. 10297-018) are from Invitrogen. Store ATP, T4 DNA ligase, and dNTPs at -20°C; when in use, keep the enzyme on ice. Return to -20°C as soon as possible.
Tris-base (Cat. No. BP152-500), Tris-Cl (Cat. No. BP152-1), boric acid (Cat. No. A74-1) and 1% ethidium bromide solution (1%) (Cat. No. BP1302-10) are from Fisher Scientific
Gel-loading solution (6x) (Cat. No. G7654; stable at 4°C), 3M sodium acetate buffer solution (Cat. No. S7899), EDTA (Cat. No. E5134), and dimethyl sulfoxide (Cat. No. D8418) are from Sigma
φ X174 RF DNA/HaeIII marker is from Promega (Cat. No. G1761)
Primer 1: 5'-AGT GGG ATT CCG CAT GCT AGT-3' containing a 5' amino linker (250 nM scale, HPLC purified) and primer 2: 5'-TAA CTA GCA TGC-3' (100nM scale, HPLC purified) are from IDT
Expand long template PCR system is from Roche (Cat. No. 1681842). Store at -20°C. Store on ice when in use; return to -20°C as soon as possible.
Reagent reservoirs with divider (Cat. No. 8095) and without divider (Cat. No. 8093) are from Matrix Technologies Corp.
10x Taq buffer II containing no MgCl2, MgCl2 (25 mM), and AmpliTaq Gold DNA polymerase (5 U/µl) are from PE Applied Biosystems (Cat. No. N8080245). Store at -20°C when in use keep the enzyme on ice. Return to -20°C as soon as possible.
Ethyl alcohol (200 proof) is from Rossville Gold Shield Chemical Co.
Adjustable single channel pipettors are from Rainin (Cat. No. Gilson Pipetman P20, P200, P1000) and Eppendorf (Reference series 2000, Cat. No. 022470001)
Adjustable 8- or 12-channel multichannel pipettors are from Matrix Technologies (Cat. No. 6019, 0.5- 12.5 µl; 6012, 5-250 µl; 6004, 15-1250 µl)
PCR machine capable of ramping at approximately 1.3°C is from PE Applied Biosystems (Model 9700, Cat. No. 4314878)
Electrophoresis power supplies (Cat. No. FBTIV816A) and UV transilluminator are from Fisher Scientific (Cat. No. FB-105)
Electrophoresis gel boxes are from USA Scientific (Cat. No. 3431-4000)
Fluorometer is from Bio-Rad (Versafluor, fluorometer, Cat. No. 170-2402)
Fan oven is from Techne (Model Hybridiser HB-1D)
Microwave is from General Electric
BioPrime DNA labeling systems (Cat. No. 18094011) [containing 2.5x random primers and Klenow fragments (40U/µl)], dNTP solutions (100mM each) (Cat. No. 10297-018), salmon sperm DNA (1mg/ml) (Cat. No. 15632-011), and redistilled, ultrapure formamide (for preparation of hybridization mixture) (Cat. No. 15515-026) are from Invitrogen and stored at -20°C Store Klenow fragments on ice when in use and return to -20°C as soon as possible.
Cy3 (Cat. No. PA53021)- and Cy5 (Cat. No. PA55021)-labeled dCTP (1mM) are from Amersham Pharmacia Biotech Inc. Store at -20°C.
Tris base (Cat. No. BP152-500), dextran sulfate (Cat. No. BP1585-100), SDS (Cat. No. 5525-087), glycerol (Cat. No. BP229-1), formamide (for preparation of array wash solution) (Cat. No. F84-1), coverslips (24 x 50mm) (Cat. No. 12531F), syringe (10ml) (Cat. No. 14- 823-28), monobasic monohydrate sodium phosphate (Cat. No. S369-500), dibasic heptahydrate sodium phosphate (Cat. No. S373-500), and microscope slides (Cat. No. 12-518-100B) are from Fisher Scientific.
Sephadex G-50 spin columns are from Amersham Pharmacia Biotech Inc. (Cat. No. 27-5330-01)
Human Cot-1 DNA is from Roche (Cat. No. 1581- 074). Store at -20°C.
20x SSC (Cat. No. S6639), EDTA (Cat. No. E5134), DAPI stain (1mM) (Cat. No. D9542) (store at -20°C), and 3M sodium acetate buffer solution (Cat. No. S7899) are from Sigma
Ethyl alcohol (200 proof) is from Rossville Gold Shield Chemical Co.
Rubber cement is from Office Depot (Cat. No. 129791)
Silicone gasket (Press-to -Seal) is from PGC Scientific (Cat. No. 49698)
Binder clips are from OfficeMax (Cat. No. OM-211)
PBS (pH 7.4) lacking Ca2+ and Mg2+ is from the UCSF Cell Culture Facility (Cat. No. BG200). Store at 4°C.
Nonidet P-40 is from Fluka (Cat. No. 74385)
Slides for printing arrays (Corning GAP) are from VWR (Cat. No. 18888-302)
Adjustable single channel pipettors are from Rainin (Cat. No. Gilson Pipetman P20, P200, P1000)
UV Stratalinker 2400 is from Stratagene (Cat. No. 400676)
Rocking table (Cat. No. 40000-300) and 37°C incubator (Cat. No. 35824-756) are from VWR
Stereomicroscope is from Leica (Model MZ8)
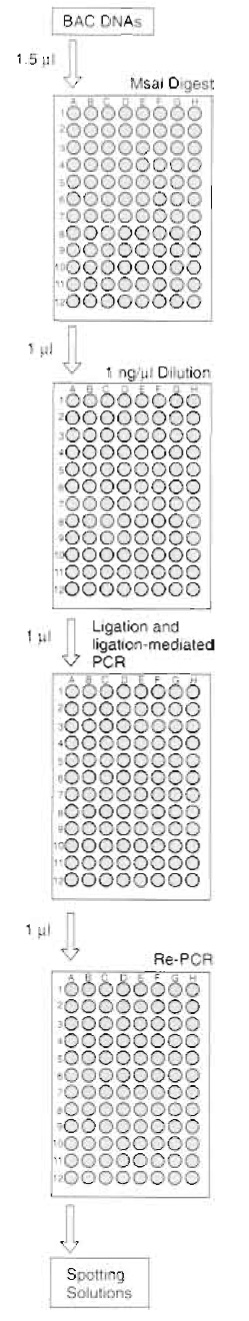 |
| FIGURE 2 Flowchart of the ligation- mediated PCR procedure for 96 BAC clones. BAC DNA (1.5 µl) is digested. One microliter of each digest reaction is diluted to 1ng/µl in H2O. Note that due to varying BAC DNA concentrations, the amount of H2O necessary to dilute 1µl of digest reaction to 1ng/µl can vary. A ligation and subsequent PCR reaction are performed using 1µl (which equals 1ng) of BAC DNA. A fraction (1 µl) of the ligation-mediated PCR is now reamplified in a second PCR reaction, which will be made into spotting solution. |
III. PROCEDURES
Procedures for the preparation of spotting solutions are described in Sections III,A-III,E and are summarized in Fig. 2. Section III,F describes the labeling and hybridization procedure.
A. Restriction Enzyme Digest of BAC DNA
The restriction enzyme digest is carried out in a 5-µl reaction volume containing 2.2x One-Phor-All Buffer Plus, 5U MseI restriction enzyme, and 20-500 ng BAC DNA.
- Restriction enzyme buffer dilution: Dilute the 10x One-Phor-All Buffer Plus to a final concentration of 0.8x in a final volume of 750 µl using sterile H2O. Dispense 93 µl into each tube of an 8-tube strip. Leftover dilution can be stored at 4°C.
- Restriction enzyme diluion: Dilute the MseI restriction enzyme to a final concentration of 5U/µl in a volume of 120µl using 10x One-Phor-All Buffer Plus. Keep both the enzyme and the dilution on ice during this process. Return the enzyme to -20°C promptly after making the dilution and discard leftover dilution.
- TBE; electrophoresis buffer: 0.089M Tris-borate, 0.089M boric acid, 0.008M EDTA. To make 10 liters, dissolve 108g Tris base, 55g boric acid, and 40ml 0.5M EDTA (pH 8.0) in distilled H2O. After dissolving, make up to 101 with distilled
H2O. Store at room temperature. - 1% agarose solution: Dissolve 1 g agarose per 100ml of electrophoresis buffer. Mix and microwave until boiling and add ethidium bromide to a final concentration of 0.5 µg/ml.
- Dispense 2.5µl of the restriction enzyme buffer dilution into each tube of twelve 8-tube PCR strips using a multichannel pipettor. Seal the tubes using a 96-well plate seal to prevent evaporation and possible contamination (see Comment 2).
- Using a single channel pipettor (see Comment 3), add 1.5µl BAC DNA (see Comment 1) to each tube. Only one 8-tube strip is handled at a time during this step. Keep the remaining eleven 8-tube strips covered using a 96-well plate seal. After adding the BAC DNA to each 8-tube strip, place the strip into a second 96-well plate rack and seal with a 96-well plate seal.
- Dispense 1 µl of the MSEI restriction enzyme dilution into each tube using a single channel pipettor. Cap each 8-tube strip using an 8-cap strip and place on ice after adding the enzyme. The restriction enzyme dilution should stay on ice during this process.
- Place the reaction into a PCR machine for an overnight incubation at 37°C, typically 12- 16h.
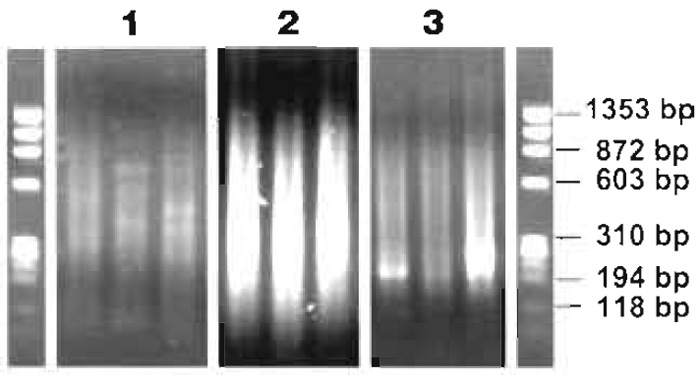
- 1.75 µl of the digest reaction can be run on a conventional 1% agarose gel containing 0.5µg/µl ethidium bromide along with a φ X174 RF DNA/HaeIII marker to check fragment length. Sizes should range from 100 to 1500bp (Fig. 3).
B. Ligation of Specific Primers to BAC DNA
The ligation reaction is carried out in a 10-µl reaction volume containing 5 µM primer 1, 5 µM primer 2, 0.5x One-Phor-All Buffer Plus, 1 mM ATP, 5 U T4 DNA ligase, and lng digested BAC DNA.
Solutions
- TE: 10mM Tris-Cl (pH 7.4), 1 mM EDTA (pH 8.0). To make 100ml, mix 1ml 1M Tris-Cl (pH 7.4) and 200µl 0.5M EDTA (pH 8.0) and make up to 100ml with distilled H2O. Autoclave to sterilize and store at room temperature.
- Primer 1: 5'-AGT GGG ATT CCG CAT GCT AGT- 3' containing a 5' amino linker. For stock solution, dissolve primer in TE (pH 7.4) to 500µM. For working solution, dilute stock solution to 100µM in distilled water. Store primer, stock solution, and working solution at -20°C.
- Primer 2: 5'-TAA CTA GCA TGC-3'. For stock solution, dissolve primer in TE (pH 7.4) to 500µM. For working solution, dilute stock solution to 100µM in distilled water. Store primer, stock solution, and working solution at -20°C.
- Ligation solution: Mix 56 µl primer 1, 56 µl primer 2, 56µl One-Phor-All Buffer Plus, and 616µl sterile water. Dispense 98 µl into each tube of an 8-tube strip. Store on ice.
- ATP solution: Dispense ~62.5µl of 10mM ATP solution into each tube of an 8-tube strip. Cap with an 8-cap strip and keep on ice when in use. Store at 4°C.
- Dilute 1 µl of each of the restriction digests to 1ng/µl using sterile H2O. Calculate the amount of sterile H2O required to dilute 1 µl of each digest to a final DNA concentration of 1ng/µl [e.g., if the BAC DNA concentration in the digest reaction is X ng/µl, the appropriate amount of sterile H2O to add is (X-1) µl] (see Comment 4). Set up twelve new 8-tube strips: individually add the calculated amount of sterile H2O to each new tube at the locations corresponding to the locations of each digest. Transfer 1µl of each digest using a multichannel pipettor. Cover using a 96-well plate seal.
- Dispense 7µl of the ligation solution onto the bottom of twelve new 8-tube strips using a multichannel pipettor (see Comment 5). Seal the 8-tube strips using a 96-well plate seal.
- Add 1 µl of the 1ng/µl BAC DNA digest to the 7 µl ligation solution in each corresponding 8-tube strip (see Comment 6).
- Place the twelve 8-tube strips into a PCR machine. Carry out the annealing reaction at 65°C for 1 min and then shift the temperature down to 15°C, with a ramp of ~1.3°C (in a Perkin-Elmer 9700 PCR machine this is a ramp rate of 5%).
- As soon as the PCR machine reaches 15°C, promptly take out the tubes from the PCR machine and carefully open all 8-tube strips, including the 8- tube strip containing the ATP solution. Using a multichannel pipettor on the repeat pipetting mode, pick up 12.5µl of 10mM ATP solution and dispense 1µl on the inside wall of each of the 96 tubes containing DNA. Gently tap the PCR rack so that the ATP slides into the DNA/primer solution. Seal with a 96-well plate seal. This procedure for adding the ATP reduces the probability of carryover contamination.
- Using a single channel pipettor, dispense 1µl of DNA T4 ligase enzyme (5U) into each of the 96 tubes.
- Place the reaction in a PCR machine for an overnight incubation at 15°C, typically 12-16h.
The ligation-mediated PCR is carried out in a reaction volume of 50 µl containing 0.6x 10xPCR buffer 1, 0.4mM dNTP mixture, 3.5U DNA polymerase mixture, and 10µl ligation mixture (Section III,B).
Solutions
- 10 mM dNTP solution: For 250 µl, mix 25 µl of each of the nucleotide stock solutions (100mM each) with 150 µl sterile H2O. Keep on ice when in use, otherwise store at -20°C.
- PCR mix: For 96 samples, mix 336µl 10x PCR buffer 1,224µl 10mM dNTP solution, and 3920 µl sterile
H2O. Mix briefly by vortexing and place on ice. - TBE; electrophoresis buffer: 0.089M Tris-borate, 0.089M boric acid, 0.008M EDTA. To make 10 liters, dissolve 108g Tris base, 55g boric acid, and 40ml 0.5M EDTA (pH 8.0) in distilled H2O. After dissolving, complete to 10 liters with distilled H2O. Store at room temperature.
- 1% agarose solution: Dissolve 1 g agarose in 100ml TBE. Mix and microwave until the boiling point has been reached and then add ethidium bromide to a final concentration of 0.5 µg/ml.
- Remove the ligations from the PCR machine. Pour the PCR mix into a reservoir. Uncap all 8-tube strips carefully. Using a multichannel pipettor on the repeat pipetting mode, pick up 205µl of the diluted dNTP mixture and dispense 40 µl on the inside wall of five subsequent 8-tube PCR strips. Purge the remaining 5 µl PCR mixture back into the reservoir. Discard the pipette tips and repeat this process. For the last two remaining 8-tube strips, pick up 85µl, while leaving the dispense volume at 40µl. Dispense 40µl on the inside wall of the two remaining 8-tube strips. Gently tap the PCR rack so that the diluted dNTP mixture slides into the DNA/primer solution. Seal with 8-tube strips.
- Place PCR tubes in the PCR machine at 68°C for 4min to melt off primer 2. After 4min, take out the tubes and add 1 µl of DNA polymerase mixture (3.5U/µl) to each tube individually using a single channel pipettor. Open one 8-tube strip at a time and close immediately after adding the enzyme mix and place on ice (see Comment 7).
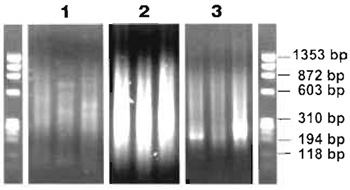
FIGURE 3 Agarose gel analysis of ligation-mediated PCR process. (1) Typical size range and DNA distribution of three MseI digest reactions. (2) Three ligation-mediated PCR products. (3) Three Re-PCR reactions. All samples were run on a conventional 1% agarose gel containing 0.5µg/µl ethidium bromide together with a φ X174 RF DNA/HaeIII DNA marker, which has a size range from 72 to 1353 bp. - Place the 8-tube strips in the PCR machine: 68°C for 3min; 94°C for 40s, 57°C for 30s, 68°C for 1 min 15s for 14 cycles, 94°C for 40s, 57°C for 30s, 68°C for 1 min 45 s for 34 cycles and 94°C for 40s, 57°C for 30s, and 68°C for 5 min for the last cycle, followed by incubation at 4°C (see Comment 8).
- Run 3.5µl of each PCR on a conventional 1% agarose gel containing 0.5µg/ml ethidium bromide along with a φ X174 RF DNA/HaeIII marker to check fragment length. Size of the PCR product should range from 70 to 2000bp, with the highest concentration of product around 200 to 800 bp (Fig. 3) (see Comment 9).
Ligation-mediated PCR is used as a template in a Re-PCR reaction to generate DNA for spotting. Amplification is carried out in a 100-µl reaction containing 1×Taq buffer II, primer 1 (4 µM), dNTP mixture (0.2 mM), MgCl2 (5.5mM), 2.5U AmpliTaq Gold, and ligationmediated PCR product (1 µl).
Solutions
- 25mM dNTP solution: For 250µl, mix 62.5µl of each of the nucleotide stock solutions (100mM each). Keep on ice when in use, otherwise store at -20°C.
- Primer 1: 5'-AGT GGG ATT CCG CAT GCT AGT- 3' containing a 5' amino linker. For stock solution, dissolve primer in TE (pH 7.4) to 500µM. For working solution, dilute stock solution to 100µM in distilled water. Store primer, stock solution, and working solution at -20°C.
- PCR mix: For 96 samples, mix 1000µl 10x Taq buffer II, 400µl primer 1 (100µM), 80µl dNTP solution (25mM), 2200 µl MgCl2 (25mM), 50 µl AmpliTaq Gold (5U/µl), and 6170µl sterile H2O. Mix briefly by vortexing and place on ice.
- TBE; electrophoresis buffer: 0.089M Tris-borate, 0.089M boric acid, 0.008M EDTA. To make 10 liters, dissolve 108g Tris base, 55g boric acid, and 40ml 0.5M EDTA (pH 8.0) in distilled H2O. After dissolving, make up to 10 liters with distilled H2O. Store at room temperature.
- 1% agarose solution: Dissolve 1 g agarose in 100 ml TBE. Mix and microwave until the boiling point has been reached and then add ethidium bromide to a final concentration of 0.5 µg/ml.
- Pour the PCR mix into a reservoir. Using a multichannel pipettor, dispense 99µl into each tube of twelve 8-tube strips. Seal using a 96-well plate seal.
- Add 1 µl ligation-mediated PCR product to each corresponding 8-tube strip using a multichannel pipettor. Do this by unsealing one 8-tube strip of PCR mixture and one 8-tube strip containing the ligationmediated PCR. Cap the 8-tube strip after adding the template and then cap the 8-tube strip containing the remaining ligation-mediated PCR. Move on to the next 8-tube strip.
- Place the 8-tube strips, now containing 99 µl PCR mixture and 1µl ligation-mediated PCR product, in a PCR machine: 95°C for 10min; 45 cycles at 95°C for 30 s, 50°C for 30 s, 72°C for 2 min and a final extension at 72°C for 7 min, followed by an incubation at 4°C.
- Run 4µl of each reaction on a conventional 1% agarose gel containing 0.5µg/ml ethidium bromide along with a φ X174 RF DNA/HaeIII marker to check fragment length. Size of the PCR product should range from 200 to 1500bp (Fig. 3) (see Comment 10).
E. Preparation of Spotting Solutions from Re-PCR Used for Array CGH
Steps
- Dry down the Re-PCR products to a volume of approximately 50µl by uncapping the 8-tube strips and placing the PCR rack face up in a hybridization oven set at 45°C (takes approximately 75 min). If you are using an oven in which the heat inlet is biased toward one side of the PCR rack, the rack should be rotated every 15min to allow even evaporation. Reducing the volume of the Re-PCR is necessary to accommodate the ethanol and sodium acetate for DNA precipitation.
- Precipitate the PCR products by adding 130µl prechilled 100% ethanol and 5µl 3M sodium acetate using a multichannel pipettor. Cap the 8-tube strips and invert the rack several times.
- Chill the PCR rack at -20°C for 15 min.
- Spin the plate at 1699g for 90min at 4°C.
- Remove the supernatant carefully using a multichannel pipettor and add 100µl 70% ethanol to the pellet using a multichannel pipettor and then recap the tubes.
- Vortex each 8-tube strip until the pellets come loose. After vortexing all 8-tube strips, centrifuge rack at 1699g for 45 min at 4°C.
- Remove as much of the 70% ethanol as possible using a multichannel pipettor and dry the pellet at room temperature for approximately 90min. The time depends on the amount of ethanol left on the pellet. Be careful not to overdry the pellets.
- Add 15 µl of a 20% DMSO solution to the pellets using a multichannel pipettor. Handled only one 8- tube strip at a time at this stage. Cap the tubes and resuspend by flicking the bottom of the PCR tubes to loosen the pellet and briefly centrifuge.
- Store the DNA solution at 4°C for an overnight (or longer) incubation. Use a multichannel pipettor to mix the solution until the pellet is completely in suspension. DNA solutions can be stored at 4°C (see Comment 11).
Random-primed labeling is carried out in a 50-µl reaction volume containing 200-300ng genomic DNA, 1× random primers, 40U Klenow DNA polymerase, Cy3- or Cy5-1abeled dCTP (40µM), and 1× dNTP mixture. The random-primed DNA product will typically range in size from 200 to 1500 bp, with the highest concentration around 400bp.
Solutions
- Tris base: For 1 liter of 1M Tris, dissolve 121.1g Tris base in 800ml of H2O. Adjust the pH to 7.4 with concentrated HCl. Make up the volume to 1 liter with H2O. Sterilize by autoclaving.
- EDTA: For 1 liter 0.5M EDTA, add 186.1 g of disodium ethylenediaminetetraacetate.2H2O to 800ml of H2O. Stir vigorously and adjust the pH to 8.0 with NaOH. Sterilize by autoclaving.
- 10x dNTP mixture: 2 mM each of dATP, dTTP, and dGTP, 0.5mM dCTP, 10mM Tris base (pH 7.6), and 1mM EDTA. For 200µl 10x dNTP mixture, mix 4µl each of 100 mM dATP, dGTP, and dTTP, 1 µl of 100 mM dCTP, 2 µl of 1M Tris, 0.4 µl of 0.5M EDTA, and 184.6 µl of dH2O.
Steps
- Mix ~200-300ng of genomic DNA with 20µl of 2.5x random primer solution and make up the volume to 42 µl with sterile H2O.
- Denature the DNA by heating the mixture at 99°C in a PCR machine for 10min. Briefly centrifuge and place on ice.
- Add 5 µl of the 10× dNTP mixture, 2 µl of Cy3- or Cy5-1abeled dCTP (1 mM), and 1 µl Klenow DNA polymerase (40U/µl). Mix well, briefly centrifuge, and place tube in a PCR machine at 37°C for an overnight incubation.
- Place the Sephadex G-50 column in a 1.5-ml tube and prespin the column at 730g for 1 min.
- Place the column in a clean 1.5-ml tube; apply the sample onto the column and spin at 730g for 2min to remove unincorporated nucleotides from the DNA mixture (see Comment 12).
Solutions
- Hybridization mixture: Distribute 1g of dextran sulfate (sodium salt, 500,000MW) over the entire length of a 15-ml tube. While holding the tube horizontally, squirt in 5 ml of formamide. Close the tube and shake vigorously for 30 s. Add 1ml of 20x SSC and shake vigorously for 30s. Dissolve overnight at room temperature. Add sterile H2O to a final volume of 7 ml. The hybridization mixture should be stored in aliquots at -20°C.
- Formamide wash solution: 50% formamide, 2x SSC in H2O. For 50ml, mix 25 ml formamide, 5 ml 20x SSC, and 20ml H2O, pH 7.0. Prepare fresh.
- PN buffer wash solution: 0.1M sodium phosphate, 0.1% Nonidet P-40, pH 8.0. Prepare 16 liters of 0.1M Na2HPO4 and 0.1% Nonidet P-40, pH 9. While continuously measuring the pH, adjust the pH to 8.0 with 0.1M NaH2PO4 and 0.1% Nonidet P-40 (approximately 1 liter). Make sure not to go below pH 8.0. Store at room temperature.
- DAPI solution: 90% glycerol, 10% PBS, 1µM DAPI. For 1ml, mix 900µl glycerol, 100µl PBS, and 1 µl (1mM) DAPI. Vortex and store at -20°C.
- Expose a printed array to 260,000 µJ of UV using a Stratalinker (see Comments 13 and 14).
- Fill a 10-ml syringe with rubber cement and fit a 200-µl pipette tip on the syringe outlet. Apply a rubber cement ring around the array using a stereomicroscope to observe the area of the array. Air dry the rubber cement and apply a second thick layer of rubber cement on top of the first layer. Air dry the rubber cement.
- Preparation of samples for hybridization to a ~12-mm square. Volumes should be increased for larger area arrays.
- Combine 50µl labeled test genomic DNA, 50 µl labeled reference genomic DNA, and 35 µg (measured by fluorometry) of human Cot-1 DNA. Precipitate the DNA sample mixture with ethanol by adding 2.5 volumes of icecold 100% ethanol and 0.1 volumes of 3M sodium acetate (pH 5.2). Mix the solution by inversion and collect the precipitate by centrifugation at 14,000rpm for 30min at 4°C.
- Carefully aspirate and discard the supernatant. Wipe excess liquid from the tube with a paper tissue, being careful not to disturb the pellet. Air dry the pellet for approximately 5-10min. Dissolve the pellet in 5µl sterile H2O, 10µl 20% SDS, and 35µl hybridization mixture (see Comment 15).
- Preparation of array blocking solution:
- Precipitate 50µl of salmon sperm DNA (10mg/ml) by adding 2.5 volumes of ice-cold ethanol and 0.1 volume of 3 M sodium acetate (pH 5.2). Mix the solution by inversion and collect the precipitate by centrifugation at 6400 rpm for 1-2 min.
- Carefully aspirate and discard the supernatant. Wipe excess liquid from the tube with a paper tissue, taking care not to disturb the pellet. Air dry the pellet for approximately 5-10min. Dissolve the pellet in 5µl sterile H2O, 10µl 20% SDS, and 35µl hybridization mixture (see Comment 15).
- Denature the DNA sample solution at 73°C for 10-15min and incubate at 37°C for ~60min.
- Place a silicone gasket around the rubber cement and apply 50µl of the salmon sperm DNA blocking solution to each array inside the rubber cement ring. Place a clean microscope slide on top to prevent evaporation (Fig. 4). Incubate at room temperature for 30min.
- Carefully aspirate ~30-40µl of salmon sperm hybridization mixture from the array and quickly apply the hybridization solution onto the array (see Comment 16).
- Place a microscope slide on top of the gasket and clamp the assembly together (Fig. 4). Incubate the array for 1-2 nights at 37°C on a slowly rocking table.
- Disassemble the slide assembly and rinse the hybridization solution from the slide under a stream of PN buffer.
- Wash the slides in formamide wash solution (50% formamide, 2x SSC, pH 7) for 15min at 45°C followed by a 15-min wash in PN buffer wash solution at room temperature.
- Carefully remove the rubber cement with forceps while keeping the array moist with PN buffer.
- If desired, arrays may be mounted in a DAPI solution to stain the array spots. Otherwise, rinse with sterile H2O and allow to air dry.
- Arrays are ready for imaging (see Comment 17).
- BAC DNS is purified from 25-ml cultures using a modification of the Qiagen plasmid minipurification protocol. The volumes of buffers P1, P2, AND P3 are increased from 0.3 to 1.5ml. After adding buffer P3, incubate on ice for 10min. Centrifuge at 4000 rpm for 45-60min at 4°C. Filter the supernatant through a 35-µm nylon mesh (available from small parts Inc.) prior to loading onto Qiagen-tip 20 columns. Heat buffer QF to 65°C prior to adding to the column. After elution, add 0.56µl of isopropanol to the DNA, mix, and store at 4°C overnight. Mix and precipitate the DNA for 45 min at 14,000rpm at 4°C. Air dry the pellet for 15-30min. Then dissolve in 50µl sterile H2O. The BAC DNA concentration usually ranges from 20 to 400ng/µl.
- Roll back the plate seal of the 96-well plate (host plate) conttaining the digest buffer solution until the first 8-tube strip is exposed. Take out the first 8-tube strip and add BAC DNA to each tube. Place this 8-tube strip in a clean 96-well plate (recipient plate) and place a 96-well plate seal on top of the plate so that it covers the whole plate (one end of the plate seal will cover the 8-tube strip, the other end of the seal will stick to the 96-well plate). g0 back to the host plate and roll back the plate seal until the next 8-tube strip becomes exposed. Take out the strip and add BAC DNA to each tube. Partially remove the 96-well plate seal of the recipient plate while leaving the first 8-tube strip covered. While holding the 96-well plate seal in one hand, place the next 8-tube strip in the recipient plate. Reseal the recipient plate and repeat this procedure until all 8-tube strips are done.
- The use of a multichannel pipettor for pipetting small amounts (~1µl) of enzyme has not been reliable. We found that by pipetting the enzymes MseI, T4 DNA ligase and the DNA polymerase mix separately using a single channel pipettor directly into the liquid in each tube has led to more reliable results. When the use of a multichannel pipettor is suggested, it is state in the protocol.
- To obtain accurate measurements of DNA concentration, a fluorometer should be used rather than a spectrophotometer.
- Use a 12.5-µl multichannel pipettor to pick up 8µl, dispense 7µl, and throw away the remaining 1µl and the pipette tips. The use of new pipette tips for each 8-tube strip is encouraged to ensure accuracy.
- First carefully unseal the BAC DNA dilution plate and then, using a multichannel pipettor, pick up 1µl of the first 8-tube strip. Unseal only the first 8-tube strip containing the primer solution, pick up the strip, and add the DNA directly into the 7-µl primer solution and cap both 8-tube strips after adding the DNA.
- The 4-min incubation at 68°C will displace primer 2 (12-mer; Tm=°C4°C). It is essential to swiftly add the DNA polymerase mixture to each 8-tube strip, close the 8-tube strip with a 8-cap strip, and place the strip-tube on ice in order to prevent significant reannealing of primer 2. The subsequent 3-min incubation at 68°C will displace the reannealed primer 2 and extend the, now free, 3' end of the digested BAC DNA. The process of taking twelve 8-tube strips out of the PCR machine, adding the enzyme mixture to each tube individually, placing the 8-tube strips on ice, and finally placing the 12 8-tube strips back into the PCR machine for amplification should take no longer than 10-15 min.
- Depending on the type of PCR machine, cycle temperatures and time can be adjusted to obtain optimal results.
- If a banding pattern appearas in the smear, the ligation and/or ligation-mediated PCR has not been successful and should be repeated. All the products should have a smear ranging from 70 to 2000bp. Any aberration from this smear, i.e., a small smear round 300-600bp or high molecular weight smear, is unacceptable and should therefore be discarded.
- The Re-PCR will most likely have the highest concentration of product around 200-400bp. The average PCR concentration should be 150ng/µl as determined with a fluorometer.
- The final concentration of the spotting solution should be approximately 0.8-1.3µg/µl. After printing spotting solution on a glass surfacee, the spots will become nearly invisible due to the lack of salt in the spotting solutions. Breathing on the glass slide will make the arrays visible for a short period of time. Printed arrays should be left at room temperature for overnight to dry spots (see Comment 14).
- The color of the DNA mixture is an indicator of the amount of labeled nucleotide incorporated during the labeling reaction.
- Place the slide(s) in the Stratalinker, arrays facing up. The arrays should be given a fixed amount of energy (260,000b t J) instead of the other available options the Stratalinker might have, such as autocrosslink or time. Overcross-linking the slide might result in a decrease in fluorescent hybridization signal (see Comment 14).
- Depending on the type of slides used for arraying, the properties of the slide surface, as well as slide processing steps, may be different. Always follow manufacturers' recommended guidelines for handling slides. In this protocol the described slide processing steps were developed using, for example, Corning GAP slides.
- Carefully pipette 5µl sterile H2O and 10µl 20% SDS on the pellet. Incubate at room temperature for ~15 min. Add 35 µl of hybridization mixture while stirring the solution with the pipette tip. Do not dissolve the pellet by pipetting to prevent foam formation due to the presence of SDS.
- Aspirate the salmon sperm DNA solution by tilting the array at approximately 45°, allowing the solution to slide to one side of the array. Aspirate approximately 30-40 µl of salmon sperm DNA solution using a 0.5- to 10-µl pipettor set at 10µl. Discard each tip after aspirating salmon sperm DNA solution. Make sure the array does not dry out by swiftly removing the salmon sperm DNA solution and applying the sample solution. If more than one array is being hybridized on one slide, tilt the array at approximately 45°, aspirate 20 µl of salmon sperm DNA solution from array 1, aspirate 10µl salmon sperm DNA solution from array 2, aspirate another 20µl from array 1, and apply sample solution to array 1. Finally, aspirate 30 µl from array 2 and apply sample solution to array 2. Be careful not to generate bubbles when applying the sample solution to the array. This is to ensure that the microscope slide, which is placed on top of the silicone gasket (Fig. 4), does not touch the sample solution.
- Images can be acquired using commercially available CCD or laser-scanning imaging systems (e.g., an Axon scanner 4000B). Image analysis and quantification can be done using commercially available image analysis programs (e.g., Genepix from Axon) or other programs, such as UCSF Spot (Jain et al., 2002).
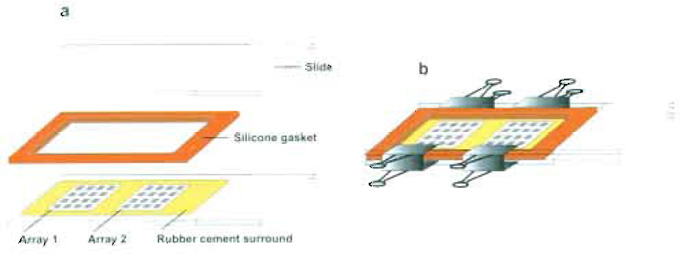 |
| FIGURE 4 Array hybridization slide assembly. A ring of rubber cement is placed closely around each array. After applying the hybridization solution to each array, a silicon gasket is placed around the arrays, partly covering the rubber cement ring. A microscope slide is placed on top of the silicon gasket, and the whole slide assembly is clamped using binder clips. (a) Individual components of the slide assembly. (b) Complete slide assembly, including binder clips. |
Jain, A. N., Tokuyasu, T. A., Snijders, A. M., Segraves, R., Albertson, D. G., and Pinkel, D. (2002). Fully automatic quantification of microarray image data. Genome Res.
Klein, C. A., Schmidt-Kittle, O., Schardt, J. A., Pantel, K., Speicher, M. R., and Riethmuller, G. (1999). Comparative genomic hybridization, loss of heterozygosity, and DNA sequence analysis of individual cells. Proc. Natl. Acad. Sci. USA 96, 4494-4499.
Pinkel, D., Segraves, R., Sudar, D., Clark, S., Poole, I., Kowbel, D., Collins, C., Kuo, W.-L., Chen, C., Zhai, Y., Dairkee, S. H., Ljung, B.-M., Gray, J. W., and Albertson, D. G. (1998). High resolution analysis of DNA copy number variation using comparative genomic hybridization to microarrays. Nature Genet. 20, 207-211.
Snijders, A. M., Nowak, N., Segraves, R., Blackwood, S., Brown, N., Conroy, J., Hamilton, G., Hindle, A. K., Huey, B., Kimura, K., Law, S., Myambo, K., Palmer, J., Ylstra, B., Yue, J. P., Gray, J. W., Jain, A. N., Pinkel, D., and Albertson, D. G. (2001). Assembly of microarrays for genome-wide measurement of DNA copy number by CGH. Nature Genet. 29, 263-264.
Solinas-Toldo, S., Lampel, S., Stilgenbauer, S., Nickolenko, J., Benner, A., Dohner, H., Cremer, T., and Lichter, P. (1997). Matrix-based comparative genomic hybridization: Biochips to screen for genomic imbalances. Genes Chromosomes Cancer 20, 399-407.
Support our developers




