Chemical Synthesis of Nucleic Acids (Oligonucleotides)
Development of strategies for chemical synthesis of nucleic acids represented a major breakthrough in molecular biology, because most of the current approaches involving PCR, manipulation of recombinant DNA, studies of gene regulation, etc. require synthetic DNA and RNA oligonucleotides with defined sequences. The difficulty of synthesizing RNA and DNA polynucleotide chains from mononucleotide units lies in the reactivity of the side chains of the bases and the susceptibility of the sugar glycosyl bond to cleavage under the harsh conditions needed for condensation reactions to generate phosphodiester bonds. An additional problem in RNA synthesis is the presence of the C´2-OH group in ribose.H. Khorana’s group was the first to solve the problem by blocking all reactive side chains of the bases with reversible blocking groups; a phosphodiester bond between C´3-OH of one nucleotide and the C´5-phosphate of another was generated by condensation in the presence of dicyclohexyl carbodiimide (DCC) under mild conditions. Repeating the process in a cyclic fashion generated oligonucleotides of a defined sequence. While the DCC condensation was efficient, the whole process was extremely laborious, because the products of each reaction had to be purified free of the side products and the blocking groups had to be removed after each cycle. Furthermore, the efficiency of the synthetic reaction fell off rapidly with increasing size of the oligonucleotide.
A major advance occurred in the 1970s when two distinct types of chemistries were invented for synthesis of deoxyoligonucleotides with the possibility of automating the cyclic procedure. Onewas based on phosphodiesters of deoxynucleotides as the starting material, which had been utilized early on for synthesis of oligodeoxynucleotides. However, the phosphoramidite method invented later has become the exclusive method of choice for synthesis of both RNA and DNA sequences. The advantages of this method are (1) the relatively high stability of the starting compounds and (2) the mild reaction conditions for removal of the protective groups.
Automated procedures have been developed for solidstate synthesis of polymers (Fig. 9), which is initiated by covalent attachment of the first monomer phosphoramidite unit to a glass matrix in the reaction vial; the phosphodiester condensation reaction is carried out by addition of monomer units in the 3´→5´ direction, which is opposite to the direction of enzymatic synthesis. Each cycle of synthesis involves removal of the protective groups after the condensation reaction. Fixed amounts of phosphoramidites of four nucleotides, as well as other modified nucleotides, are added to the reaction vial in predetermined order and amounts. The chemical treatments involving acidic and alkaline solvents are carried out in a preprogrammed sequential order, and the glass matrix containing the oligonucleotide is washed with solvent in between each reaction. The complete procedure has been automated in several commercial instruments. After synthesis is completed, the oligonucleotide product is released from the glass matrix by alkaline treatment, and then the last protective trityl group is removed. The quality and efficiency of polymer synthesis is determined by the efficiency of the individual reactions. The major advantage of phosphoramidite-based synthesis is very high efficiency (99%) of both the condensation and the deprotection reactions. Nonetheless, it is obvious that because the final yield of the oligonucleotide is the product of the yields of each individual cycle, very long oligonucleotides cannot be synthesized at a significant level. In practical terms, the current size limit of an oligonucleotide is usually up to about 120 monomer units. Even then the product has to be purified (usually by gel electrophoresis) from the contaminants, mostly composed of failed synthesis material.
A major problem in therapeutic use of oligonucleotides is their degradation by nonspecific nucleases, once delivered inside the tissues and cells. One of several approaches to counter this problem is to synthesize artificial nucleic acids in which phosphate oxygen is replaced with sulfur. In a phosphorothioate oligo (S-oligo), some or all of the internucleotide phosphate groups are replaced by a phosphothioate group. These S-oligos are widely used in anti-sense applications because of their enhanced stability. The modified backbone of an S-oligo is resistant to the action of most nucleases and endonucleases, but they also tend to be subject to more nonspecific interactions due to “stickiness.”
A. Peptide Nucleic Acids (PNA)
Peptide nucleic acids (PNA; Fig. 10) are synthetic polynucleobase molecules which bind to DNA and RNA with high affinity and specificity. PNA was constructed with a charge-neutral, achiral, pseudopeptide backbone and is therefore chemically more closely related to peptides than to nucleic acids. Thus, PNAs, because of their backbone properties, show extremely good nucleic acid hybridization properties. In fact, PNA–DNA and PNA–RNA duplexes are, in general, thermally more stable than the corresponding DNA(RNA)–DNA(RNA) duplexes.
PNAs are relatively easy to synthesize and are stable (especially biologically). These make PNA an attractive candidate for developing effective anti-sense and anti-gene reagents and drugs. PNAs have been found to inhibit RNA polymerase, human telomerase,HIVreverse transcriptase, and many more. Such PNAs are candidates for anti-cancer drugs and also as a means of developing novel drugs to treat HIV infections (AIDS). Despite these encouraging results, further progress is very much impeded by the in efficient uptake of PNA by living cells and the lack of efficient delivery systems.
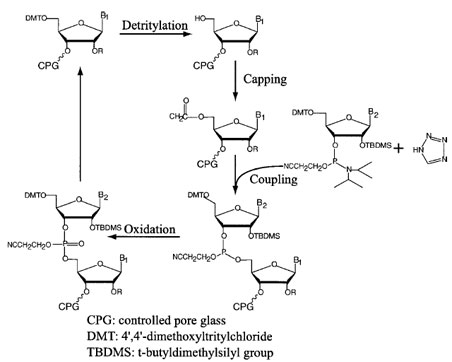 |
| FIGURE 9 An outline of the chemical synthesis of nucleic acids. |
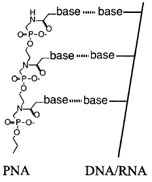 |
| FIGURE 10 Structure of peptide nucleic acid (PNA). An artificial oligomer produced by chemical synthesis retains the ability to pair with bases, but is resistant to degradation by nucleases because its backbone does not contain the normal phosphodiester linkage. |
Support our developers




