Secondary Structural Motifs
The major three-dimensional motifs found in proteins were predicted to exist by Cory and Pauling in 1951 before the first protein structure determination through their study of the structures of small peptides. They recognized that secondary structural motifs must accommodate the hydrogen bonding potential of the peptide bond as well as utilize the conformational angles found in model peptides. This emphasizes the importance of hydrogen bonds in specifying the conformation of the polypeptide chain. In general every potential hydrogen bond donor and acceptor in a protein participates in one or more hydrogen bonds. This requirement explains the common occurrence of the α-helix and β-sheet.A. α-Helix
The first secondary structural element predicted and identified in a protein was the α-helix (Fig. 5). In the α-helix the path of the polypeptide chain follows a right-handed arrangement where carbonyl oxygen of residue (i) interacts with the amide hydrogen on residue (i + 4). There are 3.6 residues per turn with a helical rise of 1.5 Å per residue which gives a helical pitch of 5.4 Å. As a consequence the side chains extend away from the helix axis every 100°. The side chains also extend toward the N-terminus of the α-helix due to the chirality of the amino acids. This is a very compact arrangement of residues that satisfies the hydrogen bonding requirements of the polypeptide chain except for the four amide hydrogens at the N-terminus and four carbonyl oxygens at the C-terminus of the helix. The lack of hydrogen bonding at the ends of an α-helix explains why they always contain more than one turn.
As indicated in the Ramachandran plot (Fig. 4), the lefthanded α-helix is an allowed conformation. It is observed occasionally in proteins, but not for an extended number of residues on account of unfavorable interactions between side chains. Residues that adopt this conformation are usually located in turns. The dominance of the right-handed helical conformation over the left-handed is a direct consequence of the L-amino acids.
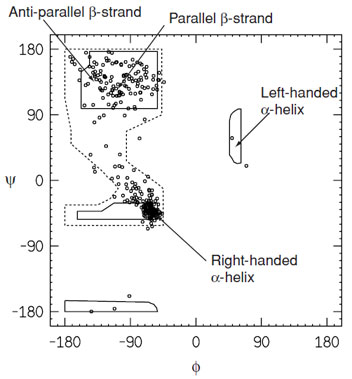 |
| FIGURE 4 Ramachandran plot for a high-resolution protein structure (human UDP-galactose 4-epimerase). This reveals that only a limited part of conformational space is occupied by the main-chain torsional angles. The space enclosed by the solid lines represents the most energetically favorable regions of conformational space. The dashed lines indicate more generously allowed regions. |
One other helical conformation is found in globular proteins: the 310 helix. This differs from the α-helix by the location of the hydrogen bond. In the 310 helix a hydrogen bond is formed between the C O(i) and H N(i+3). The packing of the main-chain atoms in the 310 helical conformation is quite tight thereby yielding nonlinear hydrogen bonds and thus is not found for extended periods. The only location that 310 helical conformation is fairly common is at the C-terminal ends of α-helices where it serves to terminate the helix with a tight turn. Three residues in the 310 helical conformation constitute a type III turn. In principle the Π-helix which has a hydrogen bond between C=O(i) and H—N(i+5) is an allowed conformation. It is not observed in proteins because the packing of the main-chain atoms would be too loose giving rise to a hole through the center of the helix. In addition there is steric hindrance between the side chains of adjacent residues along a π-helix.
B. Collagen Helix
Collagen is the most abundant protein in mammals. It is a fibrous protein that exhibits a helical repeat that is different from that of any of the previous conformations. This protein is characterized by a repeating motif (Gly—X—Y)n where X is usually proline and Y is often 4-hydroxyproline. Each collagen molecule contains ∼1000 amino acid residues and is about 3000 Å long. This protein is synthesized as a preprotein that includes 200 additional residues at both the N- and C-terminii that fold to form globular domains and serve to prevent the molecules from assembling. After post-translational hydroxylation of approximately one half of the prolines and removal of the globular extensions at both termini each collagen protomer adopts a left-handed polyproline (II) helical conformation and assembles with two other molecules to form a right-handed triple-helix (Fig. 6). Each strand of collagen is highly extended with 3.3 residues per turn and a translation of 2.9 Å per residue. Consequently individual strands are unstable and must aggregate for stability. The noninteger number of residues per turn is to accommodate the right-handed superhelix that contains 10 triplets per turn.With this arrangement, every third residue lies at the helix axis in such a way that only a glycine would fit (Fig. 6). This explains the (Gly—X—Y)n sequence motif. Interestingly there is extensive hydrogen bonding in the collagen superhelix; however, it occurs between the carbonyl oxygen on one strand with a NH on a neighboring molecule rather than within the polypeptide as seen in the α-helix. The use of proline and hydroxyproline restricts the conformational angles available to the polymer and serves to stabilize the collagen fibers. Hydroxylation of the proline increases the stability of the fibers, although the exact reason for this enhanced stability is still unclear.
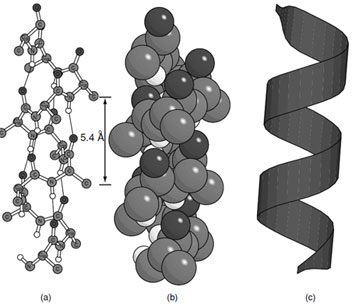 |
| FIGURE 5 The α-helix (a) ball and stick representation, (b) space filling representation, and (c) ribbon representation. |
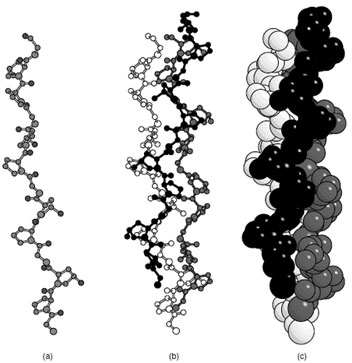 |
| FIGURE 6 The collagen helix (a) single strand of Gly–Pro–Hyp, (b) triple helix of collagen, (c) space filling representation of the collagen triple helix. |
C. β-Sheet
The second most common and identifiable secondary structural conformation is the β-strand. In contrast to the α-helix, the polypeptide chain in a β-strand is almost completely extended with a translation per residue of 3.4 Å . An isolated strand is unstable because there are no interactions between residues that are close in sequence. Thus the β-strandis onlyobservedinconjunctionwithother strands where it can form complementary hydrogen bonds with opposing peptide groups. These strands can either associate in a parallel or antiparallel manner to form β-sheets. Association of multiple strands gives rise to larger sheets that may be built from a variety of antiparallel and parallel strands; however, there is a strong tendency to prefer structural motifs that are dominated by mostly parallel or antiparallel strands.
In both antiparallel and parallel strands the peptidyl oxygen and amide hydrogen form almost ideal hydrogen bonds with neighboring strands; however, the geometry is somewhat different in each type of sheet (Fig. 7). Both arrangements lead to stable structures; however, antiparallel β-sheets are generally considered to be more stable than sheets built solely from parallel strands. As discussed later parallel sheets typically are buttressed on both sides by additional layers of secondary structure where these are usually α-helices. In contrast anti-parallel β-sheets often only require one additional layer of secondary structural elements to establish a stable fold.
The side chains in both parallel and antiparallel β-sheets extend alternatively to opposite sides of the sheet. Consequently the groups on adjacent residues within a strand do not contact each other. Rather there is considerable interaction between side chains on adjacent strands such that complementarity is observed. In addition, all β-sheets have a characteristic right-handed twist when viewed along the strand. This twist is considered to be the consequence of the interactions of the side chains with the backbone of the polypeptide chain and is thus a direct result of the chirality of the amino acids. The magnitude of the twist is somewhat variable but is usually more pronounced in antiparallel β-sheets.
There is a considerable contrast between the nature and usage of the α-helix and β-sheet. α-Helices are selfcontained secondary structural elements that may contain a substantial number of amino acid residues even in globular proteins. By comparison, β-strands typically contain three to six amino acid residues and require an adjacent strand to form a stable folding unit. Proteins that are built from α-helices usually have a very high percentage of their residues in the helical conformation (∼80%) with comparatively few devoted to connecting regions. In proteins that are dominated by β-strands, typically < 50% are in the β-conformation. This occurs because for every three to six residues in each strand there must be an equivalent number of amino acids devoted to a turn to bring the polypeptide chain back into a position where it can hydrogen bond to the same or neighboring β-strand. This emphasizes the importance of turns in protein structures.
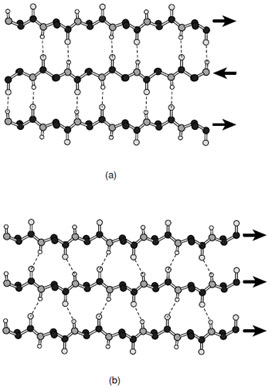 |
| FIGURE 7 Ball-and-stick representations of (a) antiparallel and (b) parallel β-sheets. By convention the direction of the polypeptide chain is taken to run from the N-terminus to the C-terminus. |
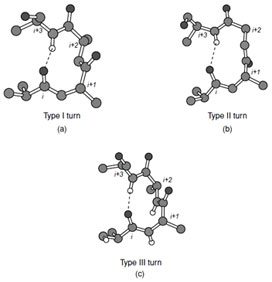 |
| FIGURE 8 Types I, II, and III turns. |
D. Turns and Random Coil
Many proteins contain secondary structure that cannot be described as either helix or turn. This is typically classified as turn, loop, or random coil. These sections of the polypeptide chain are characterized by nonrepetitive conformational angles; however, this does not necessarily imply that these residues are less stable or less well ordered than the regular secondary structural elements. Many active site residues and components critical for ligand recognition reside in loops or random coil and adopt an exquisitely well-defined conformation.
On average, one third of all residues in proteins are involved in turns that serve to reverse the direction of the polypeptide chain. These turns are an essential feature of globular proteins and are almost always located at the surface. In contrast to α-helices and β-strands which have repetitive conformational angles, the conformational angles observed in turns occur in sets that are characteristic of each type (Table II). Turns have been classified according to the commonly observed groups of conformational angles and the number of residues involved. Of these the β-hairpin or reverse turn is the most common. This type of turn is frequently used to connect antiparallel β-strands.
Three general types of reverse turn have been described; types I, II, and III, which all contain four amino acid residues and normally exhibit a hydrogen bond between C=O(i) and H=N(i+3) (Fig. 8). Of these, the type III turn consists of a short section of residues in the 310 helical conformation. Additional variants of the type I and II class are observed in the I´ and II´ turns. These exhibit conformational angles for the central two residues of the turn that are the mirror image of types I and II. The observed conformation angles favor the presence of certain amino acids at specific locations in the turns. For example, glycine predominates at position (i + 3) and proline predominates at position (i + 1) in both types I and II turns. In all turns the central two amino acid residues do not form peptidyl hydrogen bonds within the turn itself and thus must either accommodate their hydrogen bonding potential via a side chain interaction with a neighboring residue or through interactions with the solvent. Thus polar or charged residues (Asp, Asn, Ser) are often located at the first residue of the turn so that they can form a hydrogen bond to the amide hydrogen of residue (i + 2). The need to satisfy the hydrogen bonding potential of the main chain atoms accounts for the placement of most turns at the surface of the protein.
Support our developers




