Selection of Clones
To ascertain the presence or absence of tetr gene in the inserted DNA fragment in plasmid a replica plating is done from the master plate. Bacterial colonies of master plate are gently pressed with sterile velvet so that a few cells of each colony may adhere on it which is then pressed on other plate containing the nutrient medium amended with tetracycline. Plates are incubated for the growth of bacterial colonies. The appearance of colonies is compared with the master plate and those colonies that fail to grow on replica plate (Fig. 4.4E) can be said to have a plasmid which had insert DNA in the tetr gene of plasmid and had destroyed tef gene.
Colony hybridization (nucleic acid hybridization) technique
The colony hybridization technique (Fig. 4.4) is based on the availability of radioactively labeled DNA probe. A probe is radioactively labeled (P32) nucleic acid (20-40 nucleotide long) with a sequence complementary to at least one part of the desired DNA. The probe may be partially pure mRNA, a chemically synthesized oligonucleotide or a related gene, which identifies the corresponding recombinant DNA. DNA probes have commercial significance. They diagnose specific DNA sequences (genes) and are useful in the diagnosis of diseases, microbiological tests and in research as well.
Cells growing on nitrocellulose filter disc are nourished through the diffusion of nutrients from gelled nutrient medium (Fig. 4.4B). The filter disc is removed and put on blotting paper soaked with 0.5 N NaOH solution. The alkali diffuses into nitrocellulose, lyses bacterial cells and denatures their DNA. Thereafter, the filter disc is neutralized by tris (hydroxymethyl) aminomethane-HCl buffer by keeping high salt concentrations. This results in binding the DNA with nitrocellulose disc in the same pattern as the bacterial colonies; to fix the cDNA properly the filter disc is baked at 80°C (Fig. 4.4C).
By thorough washing unhybridized (unbound) probes are removed from the hybridized probe (colonies containing sequences complementary to probe) and is identified by autoradiography of the nitrocellulose filter disc (Fig. 4.4E-F). Colonies which develop positive X-ray image (Fig. 4.4G) are compared with master plate and picked up, and multiplied on the nutrient medium.
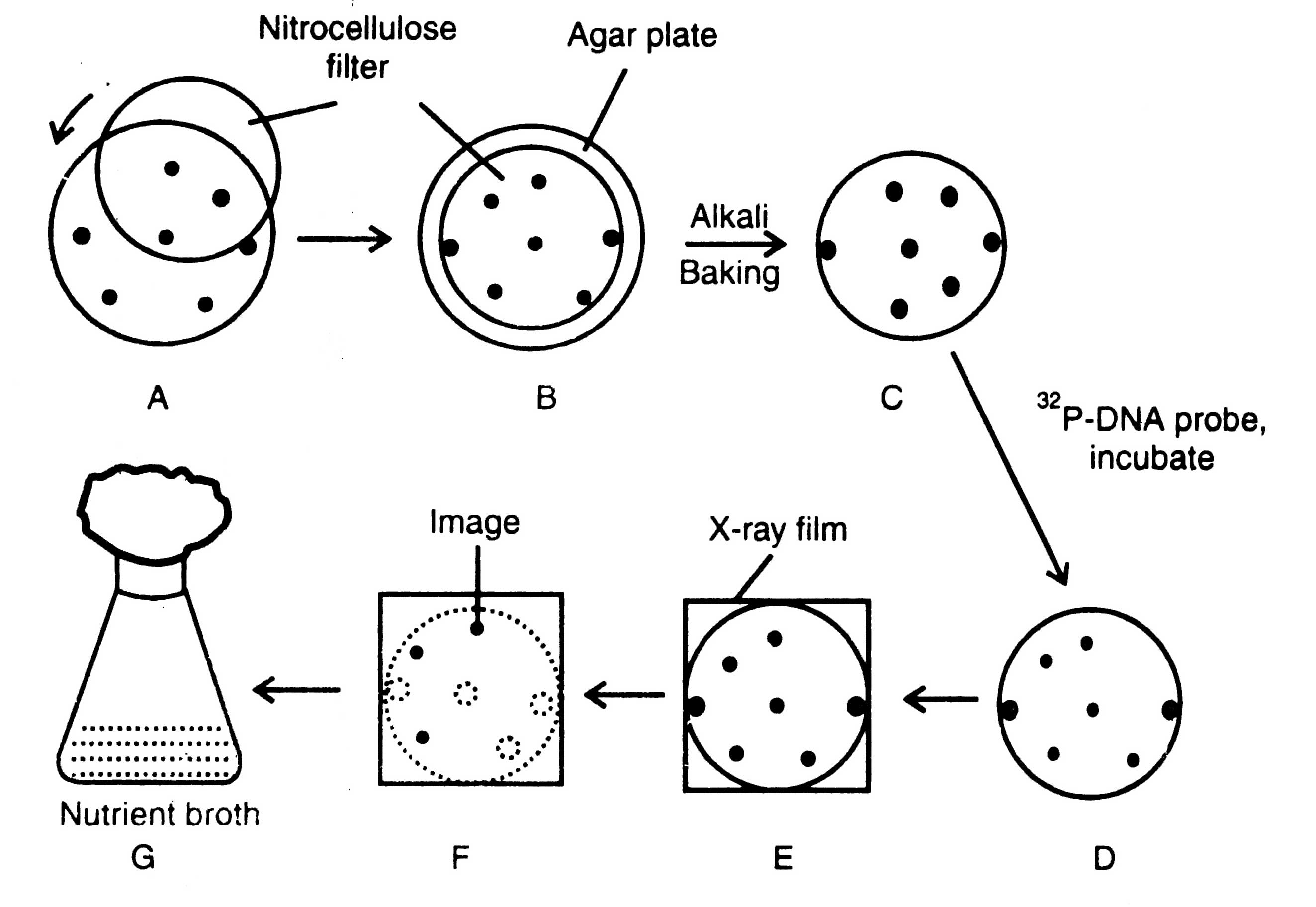
Fig. 4.4. Colony hybridization technique.
A similar procedure is followed to make replica of plaques developed in the bacterial lawn by bacteriophage recombinants. Other methods for selection of clones or screening are in vitro translation, and direct immunological techniques.
In vitro translation
In vitro translation is now used as a method to confirm the identification of recombinant clones. Nagata et al. (1980) have used primary translation screened by isolating the interferon gene from total leukocyte poly A+RNA. Translation of poly A+RNA is done when it is microinjected into oocytes from the toad, Xenopus laevis with the result of secretion of interferon into culture medium.
Immunological tests
The immunological techniques are the final test analogous to colony hybridization technique as described earlier. It is an alternative screening procedure which relies on expression, and generally applicable approach to identify a clone synthesizing a particular polypeptide. This is potentially a very powerful method since the only absolute requirement is that the required mRNA encodes a protein for which a suitable antibody is available (Williams, 1981).
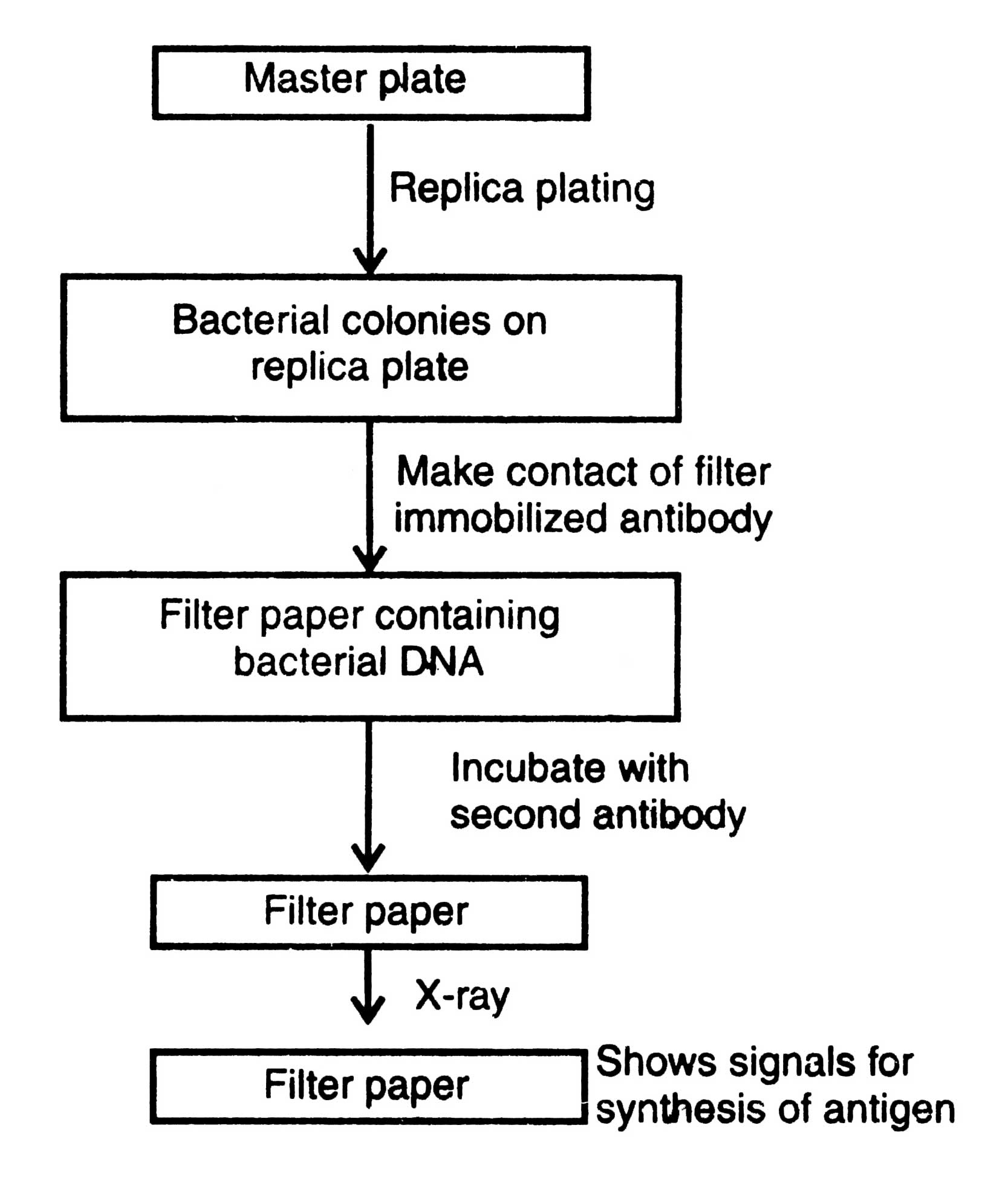
Fig. 4.5. Steps of immunological test.
Two in situ techniques given by Broome and Gilbert (1978), and Erlich et al. (1978) are in current use.
In the immunological test, instead of radio-labeling of DNA molecules, antibodies (immunoglobulins) are used to identify the colonies or plaques developed on master plates that synthesize antigens encoded by the foreign DNA present in plasmids of the bacterial clones. For this purpose a special vector, known as expression vector, is designed where the foreign DNA is transcribed and translated within the bacterial cell (Glover, 1984). The growth medium containing specific anti-serum may help in detection of viable immunoprecipitate (precipitin) around the colonies or plaques. The method follows : (i) the replica plating of bacterial colonies of master plate on nutrient agar, (ii) lysis of cells after their growth by exposure to chloroform vapor, or treatment with high temperature, (iii) making gentle contact of a solid support, for example, a cellulose filter containing immobilized antibody to solid support with the lysed cells within the colonies to allow absorption of antigen to antibody, and (iv) detection of antigen-antibody complex by incubating the cellulose filter with a radio-labeled second antibody. The antibodies that do not react are washed off and position of the antigen-antibody complex is determined by passing the filter through X-ray. It gives the signal of those bacterial cells which synthesize antigen on the master plate (Fig. 4.5).
Fig. 4.6. Detection of specific antigen synthesized by a bacterium.
Broome and Gilbert (1978) purified immunoglobulin G (IgG) and bound it to an agar plate so that the antigens released through in situ lysis of bacterial colonies can bound to the fixed antibody (Fig .4.6). The antigen is then detected by using the same IgG preparation which 4.6 is radioactively labeled. This recognizes determinants on the bound antigen. 4.6 Erlich et al. (1978) used F(ab)½ fragments derived by digestion of pepsin of the immunoglobulin, bound to either polyvinyl cellulose or diazobenzyl oxymethyl cellulose (DBMC) paper. This is incubated first with the antigen and then with undigested antiserum, FC portion of which will bind radiolabelled Staphylococcus aureus A protein.
Blotting Techniques
Southern blotting techniques
A method, developed by a molecular biologist E.M. Southern (1975) for analysing the related genes in a DNA restriction fragment is called as southern blotting technique. Southern blots can easily provide a physical map of restriction sites within a gene located normally on a chromosome, and reveal the number of copies of the gene in the genome, and the degree of similarity of the gene when compared with the other complementary genes.
The radiolabelled nucleic acid probe hybridizes the complementary DNA on nitrocellulose filter. The filter is thoroughly washed to remove the probe. The hybridized regions are detected autoradiographically by placing the nitrocellulose filter in contact with a photographic film. The images show the hybridized DNA molecules. Thus, the sequences of DNA are recognized following the sequences of nucleic acid probe.
Fig. 4.7. Procedure of Southern Blotting technique.
Northern blotting technique
Southern blotting technique could not be applied directly to the blot transfer of mRNA separated by gel electrophoresis, because RNA was found not to bind with nitrocellulose filter. Alwine et ah (1979) devised a technique in which RNA bands are blot transferred from the gel onto chemically reactive paper. An aminobenzyloxymethyl cellulose paper, prepared from Whatman filter paper No. 540 after a series of uncomplicated reactions, is diazotized and rendered into the reactive paper and, therefore, becomes available for hybridization with radiolabelled DNA probes. The hybridized bands are found out by autoradiography. Thus, Alwine's method extends that of Southern's method and for this reason it has been given the jargon term 'Northern blotting'. There is nothing northern or western like Southern.
Western blotting technique
Towbin et al. (1979) developed the western blotting technique to findout the newly encoded protein by a transformed cell. In this method radiolabelled nucleic acid probes are not used. This technique follows the following steps:
- Electrophoresis of protein in polyacrylamide gel,
- Blotting of proteins onto nitrocellulose filter paper,
- Hybridization of proteins by using radiolabelled antibodies (I123- antibodies) of known structure, and
- Detection of hybridized sequences by autoradiography.
Support our developers




