Blotting Techniques—Southern, Northern, Western Blotting
Theory: Complementarity and Hybridization
Molecular searches use one of several forms of complementarity to identify the macromolecules of interest among a large number of other molecules. Complementarity is the sequence-specific or shape-specific molecular recognition that occurs when 2 molecules bind together. For example: the 2 strands of a DNA double-helix bind because they have complementary sequences; also, an antibody binds to a region of a protein molecule because they have complementary shapes.
Complementarity between a probe molecule and a target molecule can result in the formation of a probe-target complex. This complex can then be located if the probe molecules are tagged with radioactivity or an enzyme. The location of this complex can then be used to get information about the target molecule.
In solution, hybrid molecular complexes hybrids of the following types can exist:
- DNA-DNA. A single-stranded DNA (ssDNA) probe molecule can form a double-stranded, base-paired hybrid with a sDNA target if the probe sequence is the reverse complement of the target sequence.
- DNA-RNA. A single-stranded DNA (ssDNA) probe molecule can form a double-stranded, base-paired hybrid with an RNA (RNA is usually a single-strand) target if the probe sequence is the reverse complement of the target sequence.
- Protein-Protein. An antibody probe molecule (antibodies are proteins) can form a complex with a target protein molecule if the antibody’s antigenbinding site can bind to an epitope (small antigenic region) on the target protein. In this case, the hybrid is called an “antigen-antibody complex” or “complex” for short.
There are 2 important features of hybridization:
- Hybridization reactions are specific; the probes will only bind to targets with complementary sequences.
- Hybridization reactions will occur in the presence of large quantities of molecules similar but not identical to the target. That is, a probe can find 1 molecule of target in a mixture of many of the related but noncomplementary molecules. These properties allow you to use hybridization to perform a molecular search for 1 DNA molecule, or 1 RNA molecule, or 1 protein molecule in a complex mixture containing many similar molecules.
These techniques are necessary because a cell contains tens of thousands of genes, thousands of different mRNA species, and thousands of different proteins. When the cell is broken open to extract DNA, RNA, or protein, the result is a complex mixture of all the cell’s DNA, RNA, or protein. It is impossible to study a specific gene, RNA, or protein in such a mixture with techniques that cannot discriminate on the basis of sequence or shape. Hybridization techniques allow you to pick out the molecule of interest from the complex mixture of cellular components and study it on its own.
Blots are named for the target molecule.
- Southern blot. DNA cut with restriction enzymes-probed with radioactive DNA.
Northern blot. RNA-probed with radioactive DNA or RNA.
Western blot. Protein-probed with radioactive or enzymatically tagged antibodies.
The formation of hybrids in solution is of little experimental value—if you mix a solution of DNA with a solution of radioactive probe, you end up with just a radioactive solution. You cannot tell the hybrids from the nonhybridized molecules. For this reason, you must first physically separate the mixture of molecules to be probed on the basis of some convenient parameter.
These molecules must then be immobilized on a solid support, so that they will remain in position during probing and washing. The probe is then added, the nonspecifically bound probe is removed, and the probe is detected. The place where the probe is detected corresponds to the location of the immobilized
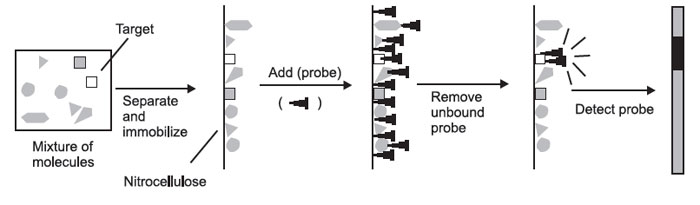 |
In the case of Southern, Northern, and Western blots, the initial separation of molecules is done on the basis of molecular weight.
In general, the process has the following steps:
- Gel electrophoresis
- Transfer to solid support
- Blocking
- Preparing the probe
- Hybridization
- Washing
- Detection of probe-target hybrids
Gel Electrophoresis
This is a technique that separates molecules on the basis of their size.
First, a slab of gel material is cast. Gels are usually cast from agarose or polyacrylamide. These gels are solid and consist of a matrix of long, thin molecules, forming submicroscopic pores. The size of the pores can be controlled by varying the chemical composition of the gel. The gel is cast soaked with buffer.
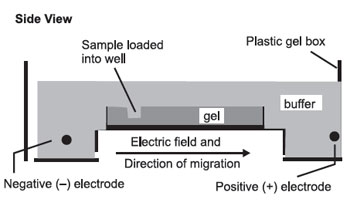
The gel is then set up for electrophoresis in a tank holding buffer, with electrodes to apply an electric field:
The pH and other buffer conditions are arranged so that the molecules being separated carry a net (–) charge so that they will be moved by the electric field from left to right. As they move through the gel, the larger molecules will be held up as they try to pass through the pores of the gel, while the smaller molecules will be impeded less and move faster. This results in a separation by size, with the larger molecules nearer the well and the smaller molecules farther away.
 |
Notes
This separates on the basis of size, not necessarily molecular weight. For example, two 1000-nucleotide RNA molecules, one of which is fully extended as a long chain (A); the other of which can base-pair with itself to form a hairpin structure
(B):
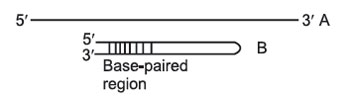 |
As they migrate through the gel, both molecules behave as though they were solid spheres whose diameter is the same as the length of the rod-like molecule. Both have the same molecular weight, but because B has secondary (2') structure that makes it smaller than A, B will migrate faster than A in a gel. To prevent differences in shape (2' structure) from confusing asurements of molecular weight, the molecules to be separated must be in a long, extended rod conformation—no 2' structure. In order to remove any such secondary or tertiary structure, different techniques are employed for preparing DNA, RNA, and rotein samples for electrophoresis.
Preparing DNA for Southern Blots
DNA is first cut with restriction enzymes and the resulting double-stranded
DNA fragments have an extended rod information without pretreatment.
Preparing RNA for Northern Blots
Although RNA is single-stranded, RNA molecules often have small regions that can form base-paired secondary structures. To prevent this, the RNA is pretreated with formaldehyde.
Preparing Proteins for Western Blots
Proteins have extensive 2' and 3' structures and are not always negatively charged. Proteins are treated with the detergent SDS (sodium dodecyl sulfate), which removes 2' and 3' structure and coats the protein with negative charges. If these conditions are satisfied, the molecules will be separated by molecular weight, with the high-molecular-weight molecules near the wells and the lowmolecular- weight molecules far from the wells. The distance migrated is roughly proportional to the log of the inverse of the molecular weight (the log of 1/MW). Gels are normally depicted as running vertically, with the wells at the top and the direction of migration downwards. This leaves the large molecules at the top and the smaller molecules at the bottom. Molecular weights are measured with different units for DNA, RNA, and protein:
DNA. Molecular weight is measured in base-pairs, or bp, and commonly in kilobase-pairs (1000 bp), or kbp.
RNA. Molecular weight is measured in nucleotides, or nt, and commonly in kilonucleotides (1000 nt), or knt. [Sometimes, bases, or b and kb, are used.] Protein. Molecular weight is measured in Daltons (grams per mole), or Da, and commonly in kiloDaltons (1000 Da), or kDa.
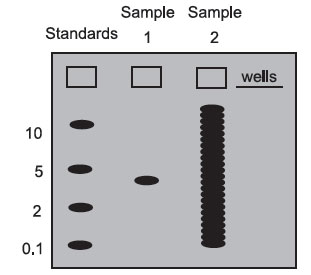
On most gels, one well is loaded with a mixture of DNA, RNA, or protein molecules of known molecular weight. These “molecular weight standards” are used to calibrate the gel run, and the molecular weight of any sample molecule can be determined by interpolating between the standards. Below is a gel stained with a dye: a colored molecule that binds to a specific class of macromolecules in a sequence-independent manner (probes bind in a sequencedependent manner).
Sample 1 contains only one size class of macromolecule—it could be a plasmid, a pure mRNA transcript, or a purified protein. In this case, you would not have to use a probe to detect the molecule of interest since there is only one type of molecule present. Blotting is usually necessary for samples that are not complex mixtures. By interpolation, its molecular weight is roughly 3.
Sample 2 is what a sample of total DNA cut with a restriction enzyme, total cellular RNA, or total cellular protein would look like in a gel stained with a sequence-independent stain. There are so many bands that it is impossible to find the one we are interested in. Without a probe (which acts like a sequencedependent stain), we cannot get very much information from a sample like this.
Top View
 |
Different stains and staining procedures are used for different classes of macromolecules:
Staining DNA. DNA is stained with ethidium bromide (EtBr), which binds to nucleic acids. The DNA-EtBr complex fluoresces under UV light.
Staining RNA. RNA is stained with ethidium bromide (EtBr), which binds to nucleic acids. The RNA-EtBr complex fluoresces under UV light.
Staining Protein. Protein is stained with Coomassie Blue (CB). The protein- CB complex is deep blue and can be seen with visible light.
Transfer to Solid Support
After the DNA, RNA, or protein has been separated by molecular weight, it must be transferred to a solid support before hybridization. (Hybridization does not work well in a gel.) This transfer process is called blotting and is why these hybridization techniques are called blots. Usually, the solid support is a sheet of nitrocellulose paper (sometimes called a filter because the sheets of nitrocellulose were originally used as filter paper), although other materials are sometimes used. DNA, RNA, and protein stick well to nitrocellulose in a sequence-independent manner.
The DNA, RNA, or protein can be transferred to nitrocellulose in 1 of 2 ways:
- Electrophoresis, which takes advantage of the molecules’ negative charge:
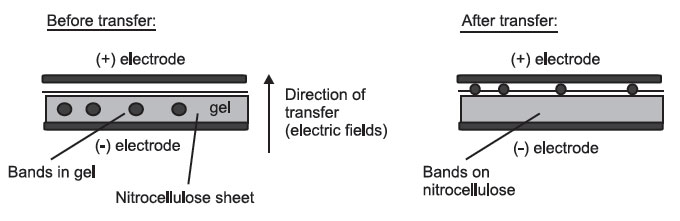 Side View
Side View
All the layers are pressed tightly together.
- Capillary blotting, where the molecules are transferred in a flow of buffer from wet filter paper to dry filter paper:
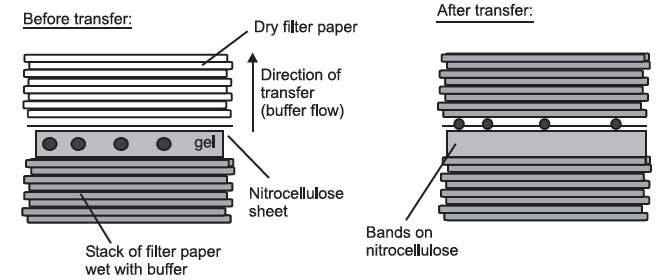 Side View
Side View
In a Southern blot, the DNA molecules in the gel are double-stranded, so they must be made single-stranded in order for the probe to hybridize to them. To do this, the DNA is transferred using a strongly alkaline buffer, which causes the DNA trands to separate. This process is called denaturation—and bind to the filter as single-stranded molecules. RNA and protein are un in the gels in a state that allows the probe to bind without this pretreatment.
Blocking
At this point, the surface of the filter has the separated molecules on it, as well as many spaces between the lanes, etc., here no molecules have yet bound. If we added the probe directly to the filter now, the probe would stick to these blank parts of the filter, like the molecules transferred from the gel did. This would result in a filter completely covered with probe, which ould make it impossible to locate the probe-target hybrids. For this reason, the filters are soaked in a blocking solution, which contains a high concentration of DNA, RNA, or protein. This coats the filter and prevents the probe from sticking to the filter itself. During hybridization, we want the probe to bind only to the target molecule.
Support our developers




