Mapping and Characterization of Protein Epitopes Using the SPOT Method
Obtaining information on the epitope recognized by a given antibody can be achieved by a variety of techniques, including antibody recognition of truncated or mutated recombinant antigens, purified antigen fragments after enzymatic digestion, or synthetic peptides predicted as being antigenic from algorithms, and so on. Methods of parallel peptide synthesis (Geysen et al., 1984) have been a breakthrough in deciphering protein-antibody interactions. By using a cleverly modified format of the standard solid-phase peptide synthesis procedure, it is possible to prepare a comprehensive set of peptides covering the entire sequence of a given protein and to probe the reactivity of the whole set of peptides in a single assay. The basic concept of parallel peptide synthesis is to divide the solid phase into discrete, addressable synthesis sites, where many different sequences can be synthesized concurrently. The initial technology made use of small polyethylene rods (called "pins"), the tips of which were chemically derivatized so as to allow peptide synthesis. Being physically separated, the pins could be used as individual chemical reactors on which peptides with defined sequences are synthesized. The pins are arrayed in a 8 × 12 format, compatible with standard ELISA plates, therefore making very practical the quantitative evaluation of peptide reactivity with a labeled antibody. The first applications of this approach were to map antibody epitopes (Geysen et al., 1984, 1986). Due to its novelty, the method stimulated research aimed at developing new strategies for parallel peptide synthesis. In 1992, the use of cellulose membranes to perform the concurrent synthesis of peptides was reported: in this approach, dubbed the SPOT method (Frank, 1992), small circular areas on a cellulose membrane are used as discrete sites for peptide synthesis.
Peptides remain attached to the membrane once the synthesis is completed, making it possible to probe the reactivity of all the peptides with a labeled ligand just by dipping the membrane into a solution of the ligand. Should any peptide be recognized by the ligand, the binding is easily revealed by appropriate means, e.g., autoradiography and enzyme-labeled secondary antibody. Once used, the membrane can be regenerated and used several times, with excellent reproducibility. The method initially described by R. Frank has been modified and improved (Molina et al., 1996; Reineke et al., 1999a; Koch and Mahler, 2002).
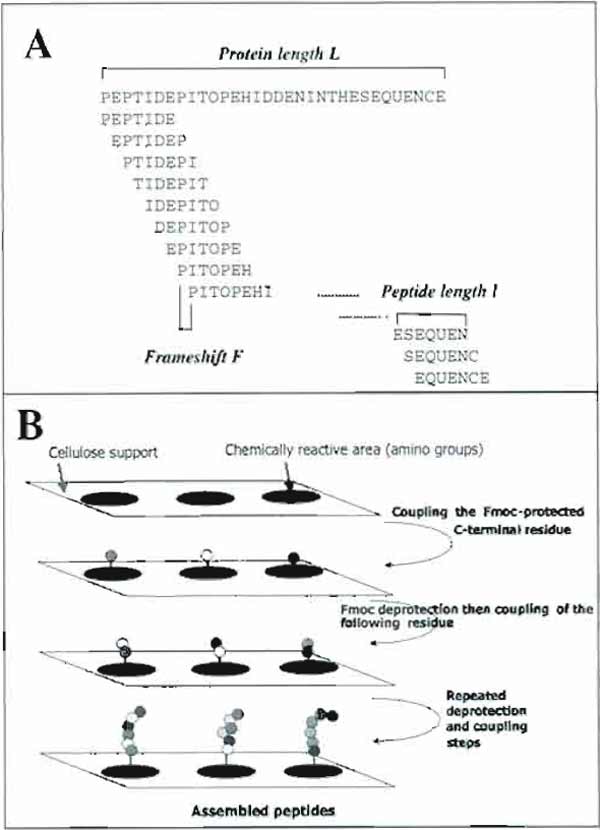 |
| FIGURE 1 Principle of epitope identification using the SPOT method. (A) Design of overlapping peptides from a putative protein sequence. (B) Principle of the SPOT method. |
However, as all methods relying on peptides, only continuous epitopes can be mapped by the SPOT approach. The identification of conformationdependent epitopes using the SPOT technique has been reported but requires slightly more sophisticated methods for revealing low-affinity binding and discarding the background signal (Reineke et al., 1999b).
The techniques used to synthesize peptides on a cellulose membrane and to probe the membrane with an antibody are described in the following sections.
A. Spotter
The ASP 222 robot from Intavis (http://www.intavis.com) is used. The protein sequence file is submitted to the specific software together with the requirements for peptide length and frameshift, and the software automatically generates the files for synthesis. The computer then drives the spotting of the activated Fmoc amino acid by the robot according to the sequence of the individual peptides. The reader is encouraged to read the instructions carefully for use of the ASP 222 robot for efficient setup of the robot. Verification of the correct functioning of the robot is recommended before starting the synthesis: a mock synthesis is initiated using a sheet of paper instead of the membrane and is then stopped once accurate spotting has occured.
B. Chemicals
Recommended most common Fmoc amino acids (from Novabiochem, http://www.merckbiosciences. de) are as follows.
Fmoc-L-Ala; Fmoc-L-Arg(Pbf); Fmoc-L-Asn(Trt); Fmoc-L-Asp(OtBu); Fmoc-L-Cys(Trt); Fmoc-LGln( Trt); Fmoc-L-Glu(OtBu); Fmoc-Gly; Fmoc-LHis( Trt); Fmoc-L-Ile; Fmoc-L-Leu; Fmoc-L-Lys(Boc); Fmoc-L-Met; Fmoc-L-Phe; Fmoc-L-Pro; Fmoc-LSer( tBu); Fmoc-L-Thr(tBu); Fmoc-L-Trp(Boc); Fmoc-LTyr( tBu); Fmoc-L-Val; Fmoc-L-Cys(Acm).
N, N'-Dimethylformamide (DMF), ref. 0343549 from SDS (http://www.sds.tm.fr/)
N-Methylpyrrolidone-2 (NMP), ref. 0873516 from SDS
Piperidine, ref. 0663516 from SDS
N, N'-Diisopropylcarbodiimide (DIPC), ref. 38370F from Sigma-Aldrich (www.sigma-aldrich.com)
N-Hydroxybenzotriazole (HOBT), ref. 02-62-0008 from Novabiochem
Methanol, ref. 20847320 from VWR (http://www.vwr.com)
Bromphenol blue, ref. B-8026 from Sigma-Aldrich
Acetic anhydride, ref. 0140216 from SDS
Trifluoroacetic acid, ref. 80203 from SDS
Dichloromethane, ref. 029337E21 from SDS
Acetic acid, ref. 20103295 from VWR
Triethylsilane, ref. 90550 from Sigma-Aldrich
Dimethyl sulfoxide (DMSO), ref. 41640 from Sigma-Aldrich
D. Membranes for Spot Synthesis
Cellulose membranes (ref. 30100) are available from Intavis (http://www.intavis.com). They can be stored for months at -20°C. They consist of cellulose paper derivatized with amino polyethylene glycol. SPOT synthesis membranes should be identified. At the end of the first synthesis cycle, when spots are colored, the first and last spot number of each row of peptides should be noted in pencil, out of the arrayed area.
A. Chemical Synthesis
Reagents
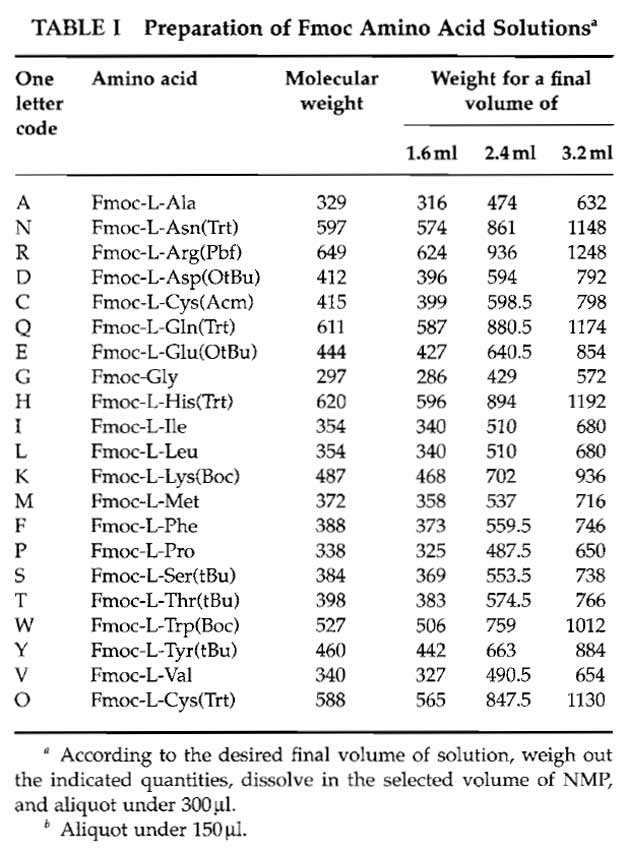
- Fmoc amino acid solutions: Weigh out each Fmoc amino acid derivative and prepare the corresponding stock solution in NMP using Table I. Use polypropylene tubes. Be sure that each derivative is completely dissolved, as at 0.6M, most of the Fmoc-protected amino acids are at their solubility limit. Distribute, in 1.5ml Eppendorf tubes, each stock solution into N 300µl aliquots except for arginine (150µl) (where N corresponds to twice the number of days the synthesis is planned to last. For example, four synthesis cycles can be performed in one working day. Supposing four membranes have to be prepared and if the peptides are 12 amino acids long, it will take 3 days to synthesize them. In this case therefore N = 8). The N series of amino acids are disposed on N tube racks and kept at -20°C.
- Bromphenol blue stock solution: Prepare 10ml of a 10mg/ml solution of bromphenol blue in pure DMF
- Activators: Prepare 1.2M HOBT (648mg/4ml NMP) and 1.2M DIPC (0.746 ml in 3.254ml NMP) and store at -20°C
- Fmoc deprotection reagent: Prepare daily 500ml of a 20% piperidine solution in DMF
- Acetylation reagent: Prepare daily 500ml of 10% acetic anhydride in DMF
- Coloration reagent: Add 5ml bromphenol blue stock solution to 395 ml DMF preface translight.
- Side chain deprotection reagent: Mix 15ml trifluoroacetic acid, 15 ml dichloromethane, and 0.75 ml triethylsilane. Warning! Trifluoroacetic acid is extremely corrosive. Gloves and protective mask should be used when preparing and using this reagent.
 |
- Purity control for DMF and NMP: Solvents used in Fmoc peptide synthesis should not contain contaminating free amines that could untimely deprotect Fmoc amino acid. To verify the absence of amines, pipette 10µl of the bromphenol blue stock solution and add 990µl of the solvent to be tested in a 1.5 ml Eppendorf tube. The solution should be light yellow. A greenish or blueish color indicates contamination with free amines, thus precluding the use of such a solvent.
- Synthesis: Each synthesis cycle involves incorporation of a defined amino acid in the sequence of all peptides at the same time. There are two coupling steps per cycle to allow the most efficient incorporation of each amino acid. For the first cycle, we recommend repeating the two coupling steps so as to ensure maximum incorporation of the C-terminal residue; it is also advisable to use a 0.2 µl spotting volume instead of 0.3 µl, which is used after.
Synthesis Steps
Thaw the membrane, activators (DIPC solution and HOBT solution), and Fmoc amino acid aliquots for 10 min before step 1.
- Activate each Fmoc amino acid aliquot: To 300µl Fmoc amino acid solution, add 150µL of 1.2M HOBT solution and 150µl 1.2M DIPC [exception: add 75µl of each solution to Fmoc-Arg (Pbf), aliquoted under 150µl. This is done because activated Fmoc-Arg (Pbf) is unstable and has to be renewed for each new cycle.]
- Place the tubes on the robot tray and initiate the synthesis cycle through the computer interface.
- Once the two coupling steps are performed, remove the membrane from the robot, place it in a polypropylene box, and then perform the following steps in a fume hood.
- Wash the membrane with DMF (3 × 2min).
- Wash the membrane with methanol (3 × 2min).
- Dry the membrane using a hairdryer on the cold position.
- Install the membrane again on the robot and initiate recoupling.
- After couplings are performed, remove the membrane from the robot.
- Wash the membrane with DMF (3 × 2 min).
- Wash the membrane with acetic anhydride 10% DMF (3 × 10min) or until the yellow color disappears.
- Wash the membrane with DMF (3 × 2 min).
- Wash the membrane with 20% piperidine in DMF (1 × 10min).
- Wash the membrane with DMF (5 × 2 min).
- Wash the membrane (2 × 2 min) with the bromphenol blue working solution (0.5ml stock solution/ 40 ml DMF).
- Wash the membrane with methanol (3 × 2min).
- Cold air dry.
- Before starting a new cycle, activate Fmoc-Arg (Pbf) as before.
- Place the membrane on the robot and initiate next cycle.
Remarks: (a) steps 4-8 are performed only for cycle 1, i.e., for incorporation of the first residue. (b) After incorporation of the last Fmoc amino acid, the deprotection step is followed by acetylation of the Nterminal residue of each peptide. The protocol is modified as follows:
- After spotting is performed, remove the membrane from the robot.
- Wash the membrane with DMF (3 × 2 min).
- Wash the membrane with 20% piperidine in DMF (1 × 10min).
- Wash the membrane with DMF (3 × 2 min).
- Wash the membrane with acetic anhydride 10% DMF (3 × 10 min).
- Wash the membrane with DMF (3 × 2 min).
- Wash the membrane with methanol (3 × 2min).
- Cold air dry.
Steps
Warning: Operate under a fume hood. The operator must wear a protection mask and gloves.
- Put the membrane into a polypropylene box.
- Add 30ml of the deprotection reagent.
- Cover the box and gently agitate it for 1 h.
- Discard the side chain deprotection reagent and wash the membrane as follows: Dichloromethane (3 × 2 min), DMF (3 × 2 min), 1% acetic acid in water (3 × 2 min), and Methanol (3 × 2 min).
- Dry the membrane with cold air.
- Place the membrane in between two pieces of filter paper and then in a sealed plastic bag and store at -20°C.
3. Cysteine pairing: Cystine-bridged peptides can be prepared on the membrane. Cysteine residues are introduced into the peptides as S-trityl derivatives. After TFA treatment, the cysteine residues are freed of their protecting group and can then be oxidized to cystine using the following procedure.
- Place the membrane in a polypropylene box.
- Add 50ml 10% DMSO in TBS.
- Cover the box and agitate it gently overnight.
- Discard the solution and wash the membrane with TBS (5 × 2min) and Methanol (3 × 2 min).
- Dry the membrane with cold air.
The length of the peptides that can be prepared in this way has theoretically no limit. However, because side products or impurities cannot be eliminated at the end of the synthesis, it is recommended to keep peptide length under 30 residues. Preparation of 12-to 15-mers is recommended for routine experiments.
If cysteine residues occur in the protein sequence, it is recommended to incorporate it into peptides as a Sacetamidomethyl cysteine derivative. This will keep the sulfhydyl groups definitively protected (to avoid unwanted reactions with the test antibodies, for example). Otherwise, substitution of Cys by Ser is also possible due to the structural similarity between both amino acids.
When preparing overlapping peptides, set the frameshift number to low values: 1 is the best, as all possible overlapping peptides are prepared, whereas a frameshift of two or three has the advantage of decreasing the total number of peptides to be prepared by a factor of two and three, respectively. The number n of overlapping peptides is related to the length of the protein L, the length of the peptides l, and the frameshift F through the formula: n = 1+[(L-l)/F]. Because there is no easy way to assess the chemical integrity of synthesized peptides, it is recommended to include in the set of peptides to be synthesized one or more a control peptides whose reactivity with a control antibody could be validated. For example, the sequence EQKLISEEDL is the epitope of the commercially available 9E10 anti-myc antibody; therefore, this peptide can be assembled on a spot and tested further for reactivity with the anti-myc antibody.
B. Epitope Identification: Membrane Probing with an Antibody
Reagents
Alkaline phosphatase-labeled species-specific antibody (preferably anti-whole molecule antibody) (e.g., anti-mouse or anti-rabbit)
Western Blocking Rangoant, ref 1921 681 from Roche Diagnostics.
MTT, ref. M2128 from Sigma-Aldrich
BCIP, ref. B6149 from Sigma-Aldrich
- Tris-buffered saline (TBS): 8g NaCl, 0.2g KCl, 6.1g Tris dissolved in 800 ml Milli-Q water. Adjust to pH 7.0 using HCl and then make up to 1 liter with Milli- Q water.
- Tween-TBS (T-TBS): Add 1 ml Tween 20 to 1 liter TBS
- Blocking buffer: 5g saccharose plus 10ml concentrated blocking buffer and 98 ml T-TBS
- Citrate-buffered saline (CBS): 8g NaCl, and 0.2g KCl, and 2.1 g citric acid monohydrate dissolved in about 800 ml Milli-Q water and adjusted to pH 7.0 using concentrated HCl. Then make the solution up to 1 liter.
- Alkaline phosphatase substrate: 180 µl MTT, 150 µl BCIP, 120µl 1M MgCl2, and 30ml of CBS
- BCIP solution: Dissolve 60mg BCIP disodium salt (Sigma) in 1 ml water. Aliquot under 150µl, keep at -20°C and in the dark.
- MTT solution: Dissolve 50mg MTT in 0.7ml DMF plus 0.3ml water. Aliquot under 200µl, keep at -20°C and in the dark.
- Regeneration reagent A: Dissolve in about 500ml Milli-Q water 480g urea and 10g sodium dodecyl sulfate (stir and gently warm). When completely dissolved, make the solution up to 1000ml. Add 0.1% 2-mercaptoethanol just before use.
- Regeneration reagent B: Mix (in this order) 500ml ethanol, 400 ml Milli-Q water, and 100 ml acetic acid.
Procedures
1. Probing the membrane: After synthesis and deprotection, the air-dried membranes should be stored in a sealed plastic bag at -20°C. Probing the reactivity of the membrane with a defined antibody takes 24 h and should be ideally started on the evening.
Prior to use, warm the membrane up to room temperature if it has been stored at-20~ and wash it three times with methanol.
- Wash the membrane three times 10min with TBS.
- Incubate overnight with blocking buffer.
- Wash with T-TBS (3 × 10min).
- Incubate with the antibody in blocking buffer (90 min, 37°C).
- Wash with T-TBS (3 × 10min).
- Add the alkaline phosphatase-labeled secondary antibody in blocking buffer and incubate for 60min at room temperature.
- Wash with T-TBS (2 × 10min).
- Wash with CBS pH 7.0. 1
1 In the case where the reactivity of spots is low, the signal can be enhanced by using 50 mM Tris buffer, pH 8.5, instead of CBS, pH 7.0. - Incubate with the alkaline phosphatase substrate (5-40 min, depending on the rapidity of apparition of the blue color on reactive spots).
- Wash with Milli-Q water (3 × 2min).
- Blocking nonspecific binding sites. The standard dilution of the concentrated blocking buffer is 1:50; however, if some background binding occurs, a more concentrated solution might be tested (e.g., 1:10).
- Choosing the appropriate dilution of the antibody. Monoclonal antibodies are generally used at 1 µg/ml in a first attempt; depending on their affinities for peptides, concentrations ranging from 0.1 to 10µg/ml can be used. We do not recommend the use of more concentrated antibody solutions, which would favor nonspecific binding. For polyclonal antibodies, a 1:1000 dilution can be tested as a first experiment.
- Detecting antibody bound to peptides. The standard procedure involves secondary antibodies coupled to alkaline phosphatase and a precipitating substrate of the latter. Although other systems can be used, this one is appreciated because of its sensitivity, very low background, and reversibility of the coloration. Stop the coloration reaction by washing the membrane (step 10) before background coloration appears.
Quantification of the results: If necessary, semiquantitative estimates of the coloration can be obtained by scanning the membrane in a black-and-white mode and using freely available software such as Scion or NIH image to integrate the resulting pixel numbers.
Regeneration of the membrane: Stripping off the membrane is important to allow reuse of the membrane. This consists in incubation with DMF to dissolve the precipitated dye and with antigen-antibody dissociation reagents. Use the following protocol.
Place the membrane into a polypropylene box and perform the successive washings in the indicated order.
- Milli-Q water (3 × 10 min).
- DMF (3 × 10min).
- Milli-Q water (3 × 10 min).
- Regeneration reagent A (3 × 10 min).
- Regeneration reagent B (3 × 10min).
- Methanol (3 × 10 min).
- Dry with cold air.
- Either start a new experiment or store the membrane at -20°C in a sealed plastic bag.
Problems and Solutions
- Lack of reactivity. There are many possible explanations for the lack of reactivity of an antibody with the membrane. The first to consider is the dependence of the antibody reactivity on protein conformation, which is often poorly mimicked by short peptides. It is nevertheless recommended to repeat the synthesis step by preparing peptides of a larger size, e.g, 25-mer peptides. This strategy has been very useful in characterizing the epitope of the antifactor VIII mouse antibody (Villard et al., 2002). There are other reasons such as low affinity of the antibody for the peptides. It is recommended to try using a more sensitive revelation system such as chemiluminescence.
- Background reactivity. The lack of cross-reactivity of the secondary antibody with peptides should be verified in a first experiment (experiment conducted in the absence of the primary antibody). Should any reactivity be observed, another secondary antibody should be tried. If many spots are colored either by the secondary antibody or by the primary plus secondary antibody, this might be due to inadequate saturation; therefore, use a more concentrated solution of blocking buffer.
- Nonremovable signal. It may occur that the reactivity of the antibody is high and that the standard procedure is not able to clean off the membrane, resulting in a "memory" signal. In this case, stronger cleaning conditions should be used in the regeneration protocol. The number of DMF washes can be increased to six and washings with reagent A and reagent B performed at 37°C. Check the membrane for the absence of reactivity in a standard experiment in which the primary antibody has been omitted.
The SPOT method of parallel peptide synthesis has proved over the years to be an excellent approach to decipher protein-protein interaction sites. Its application to the identification of epitopes recognized by monoclonal or polyclonal antibodies is one of the most direct and informative methods available. Hereafter, we briefly comment and illustrate several useful applications of the method.
Defining the main continuous antigenic regions of a protein. By using one or several polyclonal antibodies raised against the protein of interest, the SPOT method can disclose regions of the sequence that are antigenic. From the results, the corresponding antigenic peptide sequences could be made in the form of soluble synthetic peptides to be used either as an antigen to detect the antibody (typical application for diagnostics) or as an immunogen to raise site-specific antipeptide antibodies that might cross-react with the parent protein with a high probability.
Mapping the epitope recognized by a monoclonal antibody. Due to the monospecificity of monoclonal antibodies, any reactivity of an mAb with one or several peptides from a set of overlapping sequences representing collectively the protein antigen will disclose its epitope with a high degree of precision. As an example, Fig. 2 shows the straightforward identification of the epitope recognized by an anti-human factor VIII antibody.
 |
| FIGURE 2 Mapping the epitope of a monoclonal anti-human factor VIII antibody. (A) Pattern of recognition of a set of overlapping docecapeptides (frameshift 1) corresponding to (part of) the amino acid sequence of human factor VIII with an anti-factor VIII monoclonal antibody. Peptides 40 to 45 are reactive. (B) Identification of the sequence of the epitope. Sequences of peptides are aligned, and the sequence common to all reactive spots (40-45) is indicated in bold. |
It is well known that mAbs raised against globular proteins are mainly dependent on the appropriate conformation of the protein, and as such will not react with peptides. However, many other types of proteins (receptors, structural proteins, regulatory proteins) may behave differently than standard globular proteins when used as immunogens and generate peptide-reactive monoclonals.
Thanks to its versatility, the SPOT method is certainly a key technique to map peptides involved in protein interaction sites. Listing all applications of this approach is clearly beyond the scope of this article. The interested reader will find it useful to read a review on the application of methods of parallel peptide synthesis for deciphering molecular interactions in the immune system (Granier, 2002) or a more general technical book on peptide arrays (Koch and Mahler, 2002).
The authors thank Dr. Sharon Lynn Salhi for the editorial revision of the manuscript.
References
Frank, R. (1992). Spot-Synthesis: An easy technique for the positionally addressable, parallel chemical synthesis on a membrane support. Tetrahedron 48, 9217-9232.
Geysen, H. M., Meloen, R. H., and Barteling, S. J. (1984). Use of peptide synthesis to probe viral antigens for epitopes to a resolution of a single amino acid. Proc. Natl. Acad. Sci. USA 81, 3998-4002.
Geysen, H. M., Rodda, S. J., and Mason, T. J. (1986). A priori delineation of a peptide which mimics a discontinuous antigenic determinant. Mol. Immunol. 23, 709-715.
Granier, C. (2002). Special issue on "Methods of parallel peptide synthesis and their contributions to deciphering molecular interactions in the immune system." J. Immunol. Methods 267, 1-2.
Koch, J., and Mahler, M. (2002). Peptide Arrays on Membrane Supports: Synthesis and Applications. Springer-Verlag, Berlin.
Reineke, U., Kramer, A., and Schneider-Mergener, J. (1999a). Antigen sequence- and library-based mapping of linear and discontinuous protein-protein-interaction sites by Spot synthesis. Curr. Top. Microbiol. Immunol. 243, 23-36.
Reineke, U., Sabat, R., Misselwitz, R., Welfle, H., Volk, H. D., and Schneider-Mergener, J. (1999b). A synthetic mimic of a discontinuous binding site on interleukin-10. Nature Biotechnol. 17, 271-275.
Van Regenmortel, M. H., and Pellequer, J. L. (1994). Predicting antigenic determinants in proteins: Looking for unidimensional solutions to a three-dimensional problem? Pept. Res. 7, 224-228.
Villard, S., Piquer, D., Raut, S., Leonetti, J. P., Saint-Remy, J. M., and Granier, C. (2002). Low molecular weight peptides restore the procoagulant activity of factor VIII in the presence of the potent inhibitor antibody ESH8. J. Biol. Chem. 277, 27232-27239.
Support our developers




