Ribosome Display: In Vitro Selection of Protein-Protein Interactions
Ribosome display is an in vitro technology to identify and evolve proteins or peptides binding to a given target (Fig. 1) (Hanes et al., 2000a). While most selection technologies need living cells to achieve the essential coupling of genotype and phenotype, ribosome display uses the ribosomal complexes formed during in vitro translation to generate the physical coupling between polypeptide (phenotype) and mRNA (genotype) (Amstutz et al., 2001). Hence, no transformation step limiting the size of the usable library is necessary, allowing the selection from very large combinatorial libraries. In addition, the rapid selection cycles require an integral polymerase chain reaction (PCR) step, which can be used for randomization, making this method ideal for directed evolution experiments. The fact that the ribosomal complex used for selection is not covalent allows an uncomplicated separation of the mRNA from the selected ribosomal complexes, even if the selected molecules bind the target with very high affinity or are even trapped covalently (Amstutz et al., 2002; Jermutus et al., 2001). All these benefits make ribosome display a good alternative to other selection techniques, such as phage display (Smith, 1985).
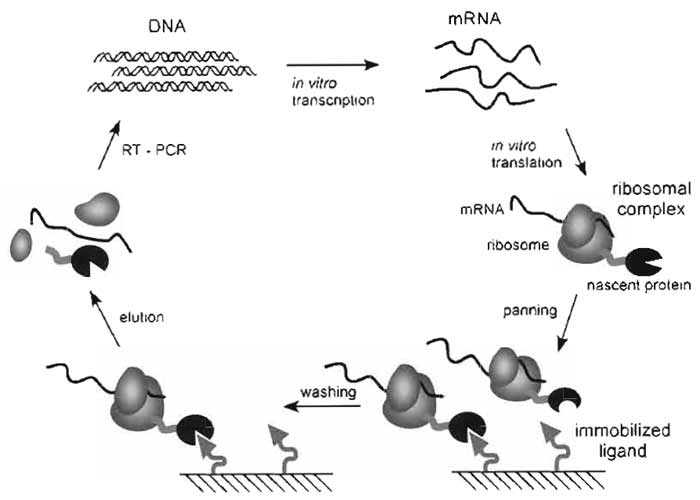 |
| FIGURE 1 Ribosome-display selection cycle. The DNA of the library of interest, fused in frame to a spacer carrying no stop codon, is transcribed in vitro. The resulting mRNA is used for in vitro translation. After a short time of translation (a few minutes) the ribosomes have probably run to the end of the mRNA and synthesized the encoded protein, but because of the absence of the stop codon, the protein remains connected to the tRNA. Stopping the translation reaction in ice-cold buffer with a high Mg2+ concentration stabilizes this ternary complex, consisting of mRNA, ribosome, and nascent protein. The spacer, occupying the ribosomal tunnel, enables the domain of interest to fold on the ribosome. These ribosomal complexes are used for affinity selection. After washing, the mRNA of the selected complexes is released by complex dissociation. The genetic information of binders is rescued by RT-PCR, yielding a PCR product ready to go for the next selection cycle. |
A. Reagents
The following chemicals and enzymes are necessary to prepare the extract and to perform ribosomedisplay selections: Luria broth base (GibcoBRL 12795- 084); agarose (Invitrogen 30391-023); glucose (Fluka 49150); potassium dihydrogen phosphate (KH2PO4, Fluka 60230); dipotassium hydrogen phosphate (K2HPO4·3H2O, Merck 1.05099.1000); yeast extract (GibcoBRL 30393-037); thiamine (Sigma T-4625); Tris (Serva 37190); magnesium acetate (MgAc, Sigma M-0631); potassium acetate (KAc, Fluka 60034); L-glutamic acid monopotassium salt monohydrate (KGlu, Fluka 49601); 20 natural amino acids (Sigma LAA-21 kit); adenosinetriphosphate (ATP, Roche Diagnostics 519 987); phosphoenolpyruvate trisodium salt (PEP, Fluka 79435); pyruvate kinase (Fluka 83328); GTP (Sigma G-8877); cAMP (Sigma A-6885); acetylphosphate (Sigma A-0262); Escherichia coli tRNA (Sigma R-4251); folinic acid (Sigma 47612); PEG 8000 (Fluka 81268); 1,4-dithiothreitol (DTT, Promega V3155); sodium chloride (NaCl, Fluka 71376); Tween- 20 (Sigma P-7949); neutravidin (Pierce 31000); bovine serum albumin (BSA, Fluka 05476), Sacchoromyces cerevisiae RNA (Fluka 83847); ribonuclease inhibitor RNasin (Promega N211B); reverse transcriptase Stratascript (50U/µl, Stratagene 600085-51); 10× Stratascript buffer (Stratagene 600085-52); dNTPs (5 mM of each dNTP, Eurogentec NU-0010-50); DNA polymerase for PCR (e.g., Vent polymerase, NEB M0254L); PCR buffer (e.g., thermopol buffer, delivered with Vent polymerase), dimethyl sulfoxide (DMSO, Fluka 41640); NTPs (50mM, Sigma); nitrocefin (Calbiochem 484400); HEPES (Sigma H-3375); spermidine (Sigma S-2501); T7 RNA polymerase (NEB M0251L); lithium chloride (LiCl, Fluka 62476); 100% ethanol (EtOH); sodium acetate (NaAc, Fluka 71180); heparin (Fluka 51550); disodium ethylenediaminetetraacetate (EDTA, Fluka 03680); T4 DNA ligase (MBI Fermentas EL0011); 4-morpholinopropanesulfonic acid (MOPS, Fluka 69949); boric acid (Fluka 15660); guanidine thiocyanate (Fluka 50990); N,N-dimethyl formamide (Sigma Aldrich 27.054-7); 37% formaldehyde (Fluka 47629); UHP water; if proteins are displayed that depend on the correct formation of disulfide bonds, protein disulfide isomerase should be used (PDI; Sigma P3818); [35S]methionine (PerkinElmer NEG009H); triethylamine (Sigma-Aldrich 90335); OptiPhase2 scintillation liquid (PerkinElmer 1200-436).
We use E. coli strain MRE600 for the preparation of the extract. This strain is RNase I deficient (Kushner, 2002) and does not contain any antibiotic resistance (Wade and Robinson, 1966).
Ribosome-display vector (pRDV), containing β- lactamase as insert (gene bank accession: AY327136).
C. Laboratory Equipment and Hardware
The following material is used in ribosome display: ART filter pipette tips (10 µl, 20 µl, 200 µl, 1000 µl, nucleic acid and nuclease-free tips, Molecular Bioproducts); QIAquick PCR purification and gel extraction kit (QIAgen 28104 and 28704); Maxisorp plate (Nunc-Immuno plate, Nunc 430341); step pipette (Eppendorf Multipipette Plus 4981 000.019) with 5- and 10-ml tips (Eppendorf 0030 069.250 and 0030 069.269); plastic seal (Corning Inc., Costar® 6524); RNase-free 1.5-ml reaction vials (MolecularBioProducts 3445); Roche high pure RNA isolation kit (Roche 1 828 655); 0.2-mm syringe filter (Millipore SLGPR25KS); dialysis tubing with a molecular weight cutoff of 6000-8000Da (e.g., Spectrum Laboratories SpectraPor 132 650).
Furthermore, standard laboratory equipment is needed, such as Sorvall RC-5C Plus centrifuge with rotors SS-34 and GS-3 or equivalent; refrigerated table centrifuge; shaker incubator; 5-liter and 100-ml baffled shake flasks for E. coli culture; Emulsiflex (Avestin, Canada) or French Press (American Instrument Company, AMINCO); 4°C room; liquid nitrogen (N2), ELISA plate shaker; UV/VIS spectrophotometer; agarose gel electrophoresis system; latex gloves, Speed-Vac (Savant Speed Vac Concentrator SVC100H); -20 and -80°C freezer; Scintillation counter.
A. Reagents
Preparation of S30 Extract
General considerations: 1 liter E. coli culture yields approximately 8 ml extract, if you plan to do ribosome display at a large scale, grow several cultures in parallel. It is important that the cells used for extract preparation are harvested in an early logarithmic phase. If the libraries used for selection contain disulfide bonds, one should omit DTT from the extract. If no disulfides need to be formed, 1mM DTT can be added to the S30 buffer as it increases the translation efficiency slightly.
Preparation of the S30 extract is performed according to Lesley, Zubay, and Pratt, with minor modifications (Chen and Zubay, 1983; Lesley, 1995; Pratt, 1984; Zubay, 1973).
Buffers
The following buffers are used in standard ribosome- display selection rounds and we advise preparing stocks: Tris-buffered saline (TBS; 50mM Tris-HCl, pH 7.4, at 4°C; 150mM NaCl), TBS with Tween [TBST; TBS with 0.05% (500µl/l) Tween-20], washing buffer with Tween (WBT; 50mM Tris-acetate, pH 7.5, at 4°C; 150mM NaCl; 50mM MgAc; 0.05% Tween-20) and elution buffer (EB; 50mM Tris-acetate, pH 7.5, at 4°C; 150mM NaCl; 25 mM EDTA); 10× MOPS (0.2M MOPS, pH 7, 50mM sodium acetate, 10mM EDTA); 10× TBE buffer (89mM Tris-buffered saline, 89mM boric acid, 10 mM EDTA).
Oligonucleotides
αtssrA: (200 µM, 5'-TTAAGCTGCTAAAGCGTAGTTTTCGTCGTTTGCGACTA-3', standard quality)
T7B: Forward RD primer. Introduces T7 promotor and part of the 5' loop
(100µM, 5'-ATACGAAATTAATACGACTCACTATAGGGAGACCACAACGG-3')
SDplus: Forward RD primer. Introduces the Shine- Dalgarno sequence and connects the T7 promoter with the FLAG tag: (100µM, 5'-AGACCACAACGGTTTCCCAATAATTTTGTTTAACTTTAAGAAGGAGATATAT
CCATGGCGGACTACAAAGATGACG-3')
tolAk: Reverse primer for RD used with tolA as spacer introducing a stabilizing 3' loop
(100µM 5'-CCGCACACCAGTAAGGTGTGCGGTTTCAGTTGCCGCTTTCTTTCT-3')
RDlinktolA: (100µM, 5'-GGGGAAAGCTTTATATGGCCTCGGGGGCCGAATTCGAATCTGGTGGCCA
GAAGCAAGCTGAAGAGGCG-3')
Primers (reverse and forward) specific for the library of interest, which must introduce appropriate restriction sites for ligation into the ribosome-display vector.
Escherichia coli strain MRE600 (Wade and Robinson, 1966); Luria broth base; incomplete rich medium: 5.6 g/liter KH2PO4, 37.8 g/liter K2HPO4·3H2O,10 g/liter yeast extract, 15 mg/liter thiaminemafter autoclaving, add 50ml 40% (w/v) glucose sterile filtered; 0.1M MgAc; 10× S30 buffer: 100mM Tris-acetate, pH 7.5, at 4°C, 140mM MgAc, 600mM KAcmstore at 4°C or chill buffer in ice bath before use; 10ml preincubation mixmmust be prepared immediately before use: 3.75ml 2M Tris-acetate, pH 7.5 (at 4°C), 71µl 3M MgAc, 75µl amino acid mix (10mM of each of the 20 natural amino acids), 0.3 ml 0.2M ATP, 0.2 g PEP, 50 U pyruvate kinase.
Material: 5-liter baffled flasks; shaker at 37°C for E. coli culture; refrigerated centrifuges (GS-3, SS-34); dialysis tubing MW cutoff 6000-8000Da; emulsiflex or French press.
Steps
Day 1
- Prepare an LB/glucose plate and streak out MRE600 on the plate. Grow it overnight at 37°C
- Prepare all chemicals, media, and buffers for E. coli extract preparation: Autoclave 1 liter incomplete rich medium, 500 ml of LB/glucose medium, and one 100-ml and one 5-liter shake flask. Prepare 50ml 40% glucose, 10ml 0.1M MgAc, and 1 liter 10× S30 buffer (use it as 1× S30 buffer afterwards). All buffers should be stored at 4°C.
Day 2
- Prepare an overnight preculture by inoculating 50ml LB/glucose medium with a colony of MRE600, which is shaken overnight at 37°C.
- Add 1 liter incomplete rich medium into the 5- liter shake flask and add 50ml 40% glucose and 10ml 0.1M MgAc both by sterile filtration (0.2-µm syringe filter).
- Inoculate the culture with 10ml overnight culture (approximately 1%) and let it shake at 37°C to OD600 = 1.0-1.2. Then transfer the culture in an icewater bath and quickly add 100 g ice (small shovel) to the culture. Shake the culture in the ice-water bath by hand for 5 min. Collect the cell pellet by centrifugation at 4°C (15 min, GS-3, 5000rpm). Wash the pellet at least three times with 50-100ml of S30 buffer.
- Determine the weight of the pellets (typically 1-1.5 g/liter). The cell pellet can now be shock-frozen in liquid nitrogen and stored at -80°C until further processing. Do not store the pellets longer than 2 days.
Day 4
Do wear gloves during all following steps!
- Thaw the pellets on ice and resuspend the cells in S30 buffer (50ml). Centrifuge at 4°C, full speed, in an appropriate centrifuge to collect the cell pellet. Resuspend the cells in 4ml/g (wet cell weight) S30 buffer.
- Lyse the cells with one passage through an EmulsiFlex (approximately 17,000psi) or a French press (at 1000psi). Repeating passages will decrease translation activity. Centrifuge the lysate at 4°C for 30min (SS-34, 20,000g). Take the supernatant and repeat this centrifugation step.
- Transfer the supernatant to a 50-ml Falcon tube. Add preincubation mix (1 ml/6.5ml supernatant) and incubate at room temperature for 60min by slowly shaking the tube. In this step most endogenous RNA and DNA will be degraded by nucleases and translatio n will run out.
- Transfer the extract into a dialysis device (MW cutoff 6000-8000Da) and dialyze the extract three times for at least 4h each at 4°C against S30 buffer (500 ml).
Day 5
- Transfer the extract into a single 50-ml tube. Aliquot the extract into RNase-free tubes immediately (e.g., 110- and 55-µl aliquots, use filter tips to avoid RNase contamination) and directly freeze the aliquots in liquid nitrogen. Store the aliquoted extract at -80°C (it will be fully active for months to years).
The premix provides the S30 extract with all amino acids (except for methionine, see later), tRNAs, the energy regeneration system, and salts, which are needed for translation. For optimal translation efficiency, every premix should be adjusted to fit the corresponding extract, especially with respect to the concentration of magnesium, potassium, PEG-8000, and amount of extract. The premix A recipe given later contains only minimal concentrations of KGlu, MgAc, and PEG-8000. By performing translations (compare Section III,F) using a test mRNA and by gradually adding increasing amounts of these components, the translation efficiency of the extract will be optimized to its maximal activity. The optimization of the premix to the $30 extract is optimally done by translating the mRNA encoding an enzyme, whose activity is determined easily, such as β-lactamase. We routinely use a cysteine-free version of the enzyme (Laminet and Plfickthun, 1989) in a ribosome-display suitable format (described in Section III,C) for optimization.
Solutions
You will need approximately equivalent amounts of premix and extract. Premix A: 250 mM Tris-acetate, pH 7.5, at 4°C, 1.75 mM of each amino acid except methionine, 10 mM ATP, 2.5 mM GTP, 5 mM cAMP, 150 mM acetylphosphate, 2.5mg/ml E. coli tRNA, 0.1mg/ml folinic acid, and 4 µM α-ssrA DNA. KGlu (180-220 mM), MgAc (5-15mM), and PEG-8000 [0-15% (w/v)] have to be adjusted to the corresponding extract. β- Lactamase assay buffer: Dissolve 5.3 mg nitrocefin in 250µl DMSO and add this to 50ml 50mM potassium phosphate buffer (pH 7) (Laminet and Pliickthun, 1989).
To avoid RNase contamination, use filter tips and wear gloves for all of the following steps.
- Mix all components to yield premix A.
- Incubate the premix in a water bath at 37°C to solubilize all components.
- Perform different in vitro translations in parallel of β-lactamase mRNA as described in (Section III,F). Add increasing concentrations of MgAc (5-15mM), KGlu (180-220mM), PEG-8000 [0-15% (w/v) of premix], and amount of extract (30-50µl for a 110-µl translation reaction).
- First optimize the MgAc concentration, then the KGlu concentration, and finally the PEG-8000 concentration. The translation time relevant for optimization should be around 10min. Stop translation by diluting the reaction five times in WBT.
- To detect β-lactamase activity, use a nitrocefin assay. Use 10-20µl of the stopped translation per milliliter β-lactamase assay buffer and follow the reaction with a photospectrometer at 486 nm.
- After determining the conditions giving the highest activity, add the chemicals at the optimal concentration to the premix A stock yielding premix Z, optimized for this very batch of extract.
- Aliquot the premix Z in RNase-free tubes and shock freeze the samples in liquid nitrogen (e.g., 500-µl aliquots).
C. Preparation of the Ribosome-Display Construct
To perform ribosome display, one needs a highquality library in the appropriate format. This section does not explain how to generate this library, as this depends entirely on the experimental goal, but rather how to convert an existing one into a format suitable for ribosome display.
Solutions, Plasmids and Strains
QIAquick PCR purification and gel extraction kit; pRDV; appropriate restriction enzymes; T4 DNA ligase.
1. Generation of the Ribosome-Display Construct via pRDV
To accelerate the procedure of bringing a library into the ribosome-display format, we generated a vector containing the necessary flanking regions (ribosome-display vector, pRDV; Fig. 2). The library is PCR amplified, cut with the appropriate restriction enzymes, and ligated into the vector such that it is in frame with the spacer (Fig. 3). A second PCR on this ligation product directly amplifies the library with all features necessary for ribosome display: the T7 promoter, the RBS, and the spacer without stop codon (Fig. 2). This PCR product is used directly for in vitro transcription to yield the library mRNA ready to go. The main advantages of the ribosome-display vector are that it can be generated in large amounts (mini to maxi prep), it is easy to handle, and always provides error-free library flanking regions. The use of the vector is not only interesting for the initial generation of the ribosome-display construct, but also for the first selection rounds. If one only amplifies the library gene of the selected clones after the panning procedure without all the flanking regions, one is able to even recover library members partly degraded by RNases in the flanking regions. The recovered genes are then religated into pRDV and one is again ready to go for another round of selection.
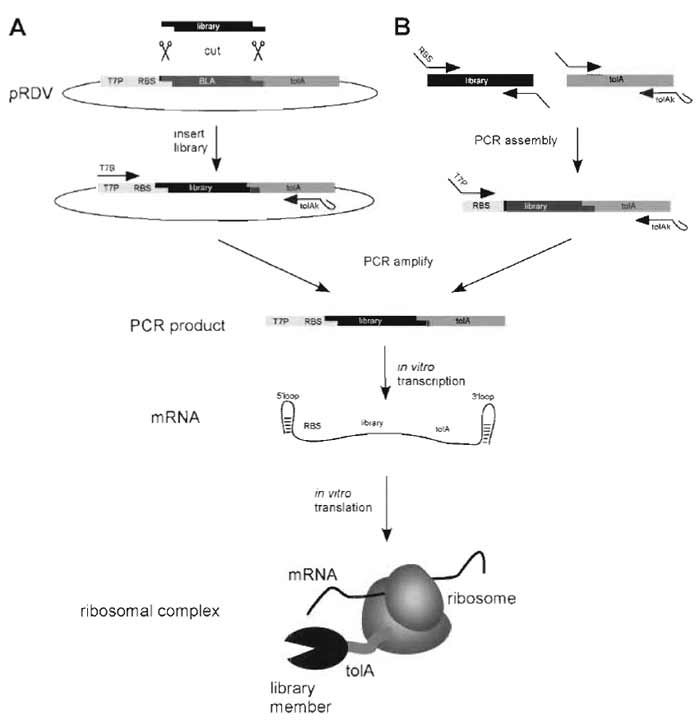 |
| FIGURE 2 Generation of the ribosome-display construct. For ribosome display the library of interest has to be flanked by an upstream promoter region and a C-terminal spacer carrying no stop codon (Fig. 3). (A) The library is PCR amplified with primers carrying restriction sites suitable for ligation into the ribosomedisplay vector (pRDV), which carries the necessary library flanking regions. The PCR product of the library is digested and ligated into pRDV. A second PCR on this ligation reaction with the primers T7B and tolAk yields a PCR product ready for in vitro transcription. (B) Alternatively, the ribosome-display construct can be generated by assembly PCR. The library and the spacer are PCR amplified separately with primers so that the C-terminal part of the library and the N-terminal part of the spacer share overlapping sequences. An assembly PCR with the library and the spacer DNA, using appropriate primers, finally yields the ribosomedisplay construct. In vitro transcription of the PCR product of either A or B yields mRNA carrying 5' and 3' stem loops (which make the mRNA more stable toward exonuclease digestion), a ribosome-binding site (RBS), the library of interest, and a spacer carrying no stop codon. By stopping the in vitro translation in icecold buffer with high Mg2+ concentration, stable ternary complexes of mRNA, ribosome, and nascent protein are formed, ready for panning. |
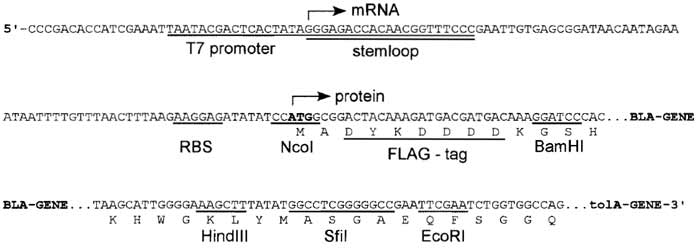 |
| FIGURE 3 DNA sequence of the expression cassette of the ribosome-display vector (pRDV). mRNA is produced from a T7 promoter starting with a 5' stem loop, with no additional overhang. The ribosomebinding site (RBS), also called Shine-Dalgarno sequence, is located upstream of the start codon. The open reading frame consists of a FLAG tag, the β-lactamase gene (serving as a dummy insert) in frame with a protein spacer, here tolA. Different restriction sites allow cloning of the library into pRDV to replace the β-1actamase gene. |
- Amplify the library with primers carrying the appropriate restriction sites for cloning into pRDV (Fig. 3).
- Digest the PCR product and purify it using QIAquick columns (≥150 ng; amount depending on the library size), pRDV is digested, optionally dephosphorylated, and the backbone is agarose gel purified. The vector insert has to be removed, as it would also ligate to the library DNA, decreasing the final complexity.
- Ligate the PCR product of the library into pRDV (molar ratio insert: vector = 7:1).
- Perform a PCR reaction on the ligation mix with the forward primer T7B (annealing on the T7 promoter with a stabilizing 5' loop) and the reverse primer tolAk (annealing on the C-terminal spacer carrying a stabilizing 3' loop).
- Analyze the DNA on an agarose gel, checking size, purity, and amount. If the band is sharp and indicates a concentration higher than 40ng/µl, the PCR product is used directly (i.e., without purification) for in vitro transcription.
2. Generation of the Ribosome-Display Construct via Assembly PCR
In some cases, it may be preferable not to ligate the library into pRDV, but to use PCR assembly to generate the ribosome display construct (Fig. 2). In this case, both the library and the spacer (e.g., tolA) are PCR amplified so that the 3' end of the library and the 5' end of the spacer share overlapping sequences (Fig. 2). The PCR products of the library and the spacer are assembled and amplified with the primers SDplus (introducing the ribosome-binding site and the connection to the T7 promoter) and tolAk. A final PCR reaction with the primers T7B (introducing the T7 promoter) and tolAk completes the construct, ready for transcription (Fig. 2).
- Amplify the library of interest with appropriate primers, introducing the FLAG tag at the 5' end of the library, on which the primer SDplus can anneal in step 3. The reverse primer should anneal on the library and add an overlap corresponding to RDlinktolA. There is no need to introduce restriction sites; however, we strongly recommend the use of the same primers as for the cloning into the RDV to have the possibility of using pRDV and to reduce the number of different fragments.
- Amplify the ribosome-display spacer using the primers RDlinktolA (forward) and tolAk (reverse). Thereby, the forward primer generates an overlap between the library and the spacer.
- In an assembly PCR reaction, the spacer is fused to the library. Mix DNA of the library with an excess of spacer DNA and perform 7 cycles of PCR without the addition of primers. For this step, we use a lower annealing temperature as for the normal amplification. After 7 cycles, add primers tolAk and SDplus to the reaction and perform another 25 cycles of amplification.
- Isolate the full-length band from an agarose gel and amplify the band using the primers T7B and tolAk.
- Analyze the DNA on an agarose gel, checking size, purity, and amount. If the band is sharp and indicates a concentration higher than 40ng/µl, the PCR product is used directly (i.e., without purification) for in vitro transcription.
D. Transcription of PCR Products
Solutions and Hardware
5× T7 polymerase buffer: 1M HEPES-KOH, pH 7.6, 150 mM MgAc, 10 mM spermidine, 200 mM DTT, NTPs (50mM each); T7 RNA polymerase; RNasin; 6M LiCl; 70% EtOH; 100% EtOH; 3M NaAc; agarose; guanidine thiocyanate; formamide; 37% formaldehyde; MOPS; Speed-Vac; heating blocks; UV/VIS spectrophotometer.
To avoid RNase contamination, use filter tips and wear gloves for all of the following steps.
- Mix the components for the following reaction using the PCR product from Section III,C. The PCR product should not be purified. Add 20.0 µl 5× T7 polymerase buffer, 14.0µl NTPs (50mM each), 4.0µl T7 RNA polymerase, 2.0µl RNasin, 22.5µl PCR product (unpurified PCR reaction), and 37.5 µl UHP water. Let the transcription reaction run for 2-3 h at 37-38°C.
- Add 100 µl UHP water and 200 µl 6M LiCl, both ice cold, and place on ice for 30 min, before centrifugation at 20,000g (4°C, 30min). Discard supernatant and wash the pellet with 500 µl ice-cold 70% EtOH. Dry the pellet on the bench (5 min, open lid) and take it up in 200µl ice-cold UHP water. Make sure it is dissolved completely before centrifuging at 20,000g (4°C, 5 min).
- Transfer 180µl supernatant to a new tube and add 20µl 3M sodium acetate and 500µl ice-cold 100% EtOH. Keep the solution on ice or at -20°C for 30min before centrifuging it at 20,000g (4°C, 30min). Discard the supernatant and wash the pellet with 500µl icecold 70% EtOH. Discard the supernatant and dry the pellet in a Speed-Vac apparatus.
- Take up the mRNA pellet in 30µl ice-cold UHP water and make sure it is dissolved completely. Take 2µl of this solution and dilute to 500µl with ice-cold UHP water for OD260 quantification and immediately N2 freeze the rest of the RNA for further use.
- Immediately (the RNA will be degraded and the signal will increase) measure the OD260. For RNA, an OD260 of 1 corresponds to a concentration of 40µg/ml.
- Add UHP water to the RNA stock in order to reach a standard concentration of 2.5µg/µl.
- The RNA quality can optionally be checked by agarose gel electrophoresis. Cast an agarose gel (1.5%, depending on your RNA size) adding 2% (w/v) of 1M guanidinium thiocyanate. Denature 5 µg (i.e., 2 µl) RNA for 10 min at 70°C in 15.5 µl sample buffer (10 µl formamide, 3.5µl 37% formaldehyde, 2µl 5× MOPS), place it on ice, add 2 µl DNA loading buffer (50% glycerol, 1 mM EDTA in H2O), and run the gel.
To perform a selection, the target molecule must be immobilized in a conformation relevant for further applications. Typically, a protein will have to be in its native conformation. A very promising way to achieve this to biotinylate the target molecule and immobilize it via neutravidin or streptavidin. Biotinylation can be done chemically with commercially available reagents, either attaching the biotin to cysteine or lysine residues. The problem of this unspecific approach is that biotinylation might destroy epitopes. Alternatively, the target protein can be expressed in recombinant form with a biotinylation tag, i.e., a peptide sequence, which is recognized and biotinylated by the E. coli biotinylation enzyme BirA (Schatz, 1993). If for any reason biotinylation is not an option, one can either immobilize the target molecule directly on the hydrophobic surface of a microtiter plate well or use a specific antibody, which itself can be immobilized easily via protein A or G. Note that the buffers used here may have to be adapted to the needs of the target molecules.
Solutions and Hardware
TBS, TBST, WBT at 4°C
Maxisorp plate; plastic seal; step pipette; plate shaker; 20µM stock neutravidin; BSA; target molecule of choice
Steps
To avoid RNase contamination, use filter tips and wear gloves for all of the following steps. We also recommend carrying out the selection, RT and PCR in duplicate, to check the reproducibility of the selection. Therefore, one target molecule is routinely immobilized in two wells.
Day 1
- Wash a Maxisorp plate three times with TBS and beat dry. Pipette (with a step pipette) 100µl of a 66 nM neutravidin solution in TBS into the wells. Seal with plastic and store overnight at 4°C. If the target molecule is not biotinylated it can also be directly immobilized by the same procedure as neutravidin, but it might denature at least partly during this procedure.
- Wash the plate incubated overnight with cold TBS three times. Add 300µl 0.5% BSA in TBS to the wells (with a step pipette) to block all hydrophobic surfaces with BSA. Seal the plate with plastic and incubate on a shaker for 1 h at room temperature. Alternatively, sensitive or fragile targets should be incubated for 2.5h at 4°C. If the target molecule is immobilized directly, incubate the BSA solution for 2h at 4°C and proceed directly to step 5.
- Wash the plate three times with TBS, beat dry, and add 200µl 0.5% BSA in TBS to each well (step pipette).
- Add 5µl of the target molecule (~10µM) to the respective well. Use a biotinylated target molecule void of free biotin. Seal with plastic and incubate on a shaker for 1 h at 4°C. To avoid binders to BSA or neutravidin, it is recommended to immobilize the target molecule only in every second well and use the alternate wells for prepanning.
- Wash the plate four times with TBS to get rid of the unbound target molecule. Wash at least once with WBT (step pipette) to equilibrate the well with the ribosome-display buffer. Incubate wells that will not be used directly for panning with WBT (amount of liquid as will be used for panning). Keep the plate at 4°C.
F. In Vitro Translation
Solutions
UHP water; WBT; WBT with 0.5% BSA; heparin (200 mg/ml); methionine (200mM); S30 extract; premix Z; the mRNA of the library; optionally PDI (4 mg/ml, reconstituted from lyophilized protein); all buffers should be kept on ice, the RNA in liquid nitrogen unless stated otherwise
We recommend carrying out the panning in duplicate to check the reproducibility of the selection. To avoid RNase contamination, use filter tips and wear gloves for all of the following steps.
- Mix the following components (amounts given are for one reaction): 13.0µl UHP water, 2.0µl Met (200mM), 41.0µl premix Z (thaw on ice, vortex before pipetting), and 50.0µl extract. Volumes might vary, depending on the batch of S30 extract. This mix can be kept on ice for a short period (few minutes). If disulfide bridges are to be formed, add 0.625 lal PDI (4 mg/ml).
- Add 4µl mRNA (2.5µg/µl) into a fresh RNasefree tube and freeze it in liquid nitrogen.
- Add the translation mix to the frozen mRNA, dissolve the pellet by flicking the tube, and translate for 6-12min at 37°C.
- In the meantime, prepare 440 µl stopping buffer [WBT with 0.5% BSA and 12.5 µl/ml heparin (200mg/ ml)] in RNase-free tubes and put them on ice.
- After the 6- to 12-min translation, pipette 100µl translation into the ice-cold stopping buffer to stop translation (always keep the ribosomal complexes on ice or at 4°C.
- Centrifuge at 20,000g, 4°C for 5 min and use the supernatant containing the ribosomal complexes for panning.
G. Panning
Solutions and Hardware
WBT; EB; yeast RNA (25µg/µl); cold room; microtiter plate shaker; Roche High Pure RNA isolation kit
All the following steps should be performed in a cold room (at 4°C or slightly below). The low temperature guarantees complex and mRNA stability. Under such conditions ribosomal complexes have survived off-rate selection procedures of 10 days and longer. During all binding and elution steps, shake the microtiter plate gently. To avoid RNase contamination, use filter tips and wear gloves.
- Add 100-200µl stopped translation mix to the well, where no target molecule is immobilized and incubate for 1 h. In this prepanning step, all BSAbinding, neutravidin-binding, or simply sticky complexes are removed. If unspecific complexes are causing problems, more than one prepanning step should be done.
- Transfer the solution to the well with the immobilized target molecule and incubate for 1 h.
- Wash the well to remove nonbinding complexes. The time span of the washing step and the number of washing steps define the selection pressure. One usually starts with short washing times in the first rounds (only rinsing six times) and increases the washing periods for later rounds (up to 3-4h). As washing is always a dilution, it is important to fill the wells to the top and exchange the buffer many times (at least six times, in all rounds!). If harsher selection pressure than simple washing is required, e.g., to select for affinities below nanomolar, one should consider immobilisation of low target molecule concentrations, competitive elution (with free target molecule in the solution), or off-rate selection procedures (see Section III,J).
- Before the mRNA of the selected complexes is eluted, prepare three RNase-free tubes for each well to be eluted: one tube with 400µl lysis buffer of the Roche RNA purification kit to purify the RNA, one tube to collect the purified mRNA, and one tube for reverse transcription. Also prepare one Roche-RNA purification column for each well to be eluted. Label all tubes and columns appropriately.
- Calculate and prepare the amount of elution buffer you need (200 µl/well) and add 50µg/ml S. cerevisiae RNA (2µl/ml of a 25-µg/µl stock) to it. Keep the buffer on ice.
- Elution is done in the cold room. Wash your well one last time with WBT, remove the supernatant completely, and beat the plate dry. Add 100µl elution buffer, shake for 10 min, transfer this eluate to the tube containing the lysis buffer, and mix well. Repeat this procedure with another 100µl of elution buffer. In the lysis buffer, the RNA is stable and can be brought to room temperature.
Solutions and Hardware
Roche High Pure RNA isolation kit; 100mM DTT; RNasin; Stratascript (50U/µl; Stratagene); dNTPs; DNA polymerase; oligonucleotides; 10× Stratascript buffer; polymerase buffer; DMSO; agarose
Steps
The RNA purification is done according to the Roche protocol, with slight modifications. All the centrifugation steps are carried out at 4°C.
mRNA Purification
- Just before RNA purification, thaw the reagents for reverse transcription (DTT, 10x Superscript buffer, dNTPs, oligonucleotide for reverse transcription).
- Set two heating blocks to 70°C and 50°C, respectively.
- Apply the lysis buffer/eluate mixture on the column and spin for I min at 8000 g. Discard the flowthrough and wash with 500µl buffer 1 (black-capped bottle in the Roche kit; the DNase incubation step, described in the Roche protocol is not necessary). Discard the flow through and wash with 500 µl buffer 2 (blue-capped bottle in the Roche kit). Discard the flow through. Add 100µl buffer 2 (blue cap) and spin for 2min at 13000g. Transfer the column to a tube to collect the RNA.
- Elute with 30µl Roche elution buffer (at 8000g for 1 min) and directly put the RNA containing collection tube to 70°C for 10min to denature the RNA. During this incubation time, pipette the reverse transcription (i.e., directly proceed with step 5).
- In a master mix tube, add the following components (amounts given are for one reaction): 0.25µl reverse primer (100µM), 0.5µl dNTP (5mM each), 0.5 µl RNasin, 0.5 µl Stratascript, 2.0 µl 10× Stratascript buffer, 2.0 µl DTT (100 mM), and 2.0 µl UHP water. Distribute the RT mix (7.75µl) to the previously labelled tubes and keep them on ice.
- Spin the denatured eluted RNA samples shortly and set them on ice. Add 12.25 µl of the eluted RNA to the RT-mix (N2 freeze the rest of the RNA directly after adding it to the reaction).
- Place the RT reaction on a 50°C heat block for 45 min.
PCR
- Mix the following components (amounts given are for one reaction): 5.0µl Thermopol buffer, 2.0µl dNTPs, 2.5µl DMSO, 1.0µl forward primer (100µM), 1.0µl reverse primer (100µM), 5.0µl RT template, 0.5 µl polymerase, and 33.0µl UHP water. PCR results can be improved if a hot start is performed (adding the polymerase in the 5-min preincubation step at 95°C).
- Run the following PCR program (to be adapted according to primers and template): 5 min at 95°C, X times (30s at 95°C, 30s at 50°C, 1 min at 72°C), 5 min at 72°C, and 4°C infinitely. Adjust the number of cycles (X = 25-45) to the corresponding selection round.
- Verify the PCR product quality by an appropriate agarose gel electrophoresis.
- Usually the quality of the PCR product is not good enough to be used directly for in vitro transcription to generate mRNA for the next round. Gel purify the desired band and perform a second PCR on the purified first PCR to yield high-quality DNA.
- If you wish to check the pool of selected clones for binders, ligate the selected library members after PCR amplification into a vector suitable for expression, transform E. coli to obtain individual clones, and test binding specificity, e.g., by crude extract ELISA. To assess the specificity of the binders, one can either compare binding of the selected molecules to the specific target molecule with the binding to an unrelated molecule or test that the specific binding can be inhibited by adding the purified unbiotinylated target molecule. The level of inhibition gives a first crude estimate of the affinity.
RT-PCR. If the RT-PCR with the forward (e.g., T7B) and reverse (e.g., tolAk) primers does not yield a high-quality product, one can amplify only the coding region of the selected library members. This usually improves the yield and the quality of the PCR product. This is most likely due to the fact that RNases degrade the mRNA from the ends. Amplifying only the central (library) stretch can rescue partly degraded clones. The PCR product of the library stretch is subsequently religated into pRDV as described in Section III,C. As this procedure rescues more clones than the PCR of the whole construct, we would recommend it for the first rounds to guarantee that no binders are lost.
Panning Controls. Ribosome display has many error-sensitive steps. It is therefore recommended to do panning, RT, and PCR in duplicate to check the reproducibility of the selection. Specific binders should be enriched from round to round. This should correlate with the number of PCR cycles needed to amplify the DNA after RT, which should decrease from round to round. Usually, one can reduce the cycle number by around five per round. If the selection pressure is increased, the yield will drop. When enrichment is observed (for designed ankyrin repeat protein libraries, (Binz et al., 2004) e.g., after round two, while for antibody libraries after round three to four), one can test the specificity of the selected pool by comparing panning results against the correct and an unrelated target molecule. If the pool is specific, only the correct target molecule will give a PCR product after RT. If the pool also gives a signal with the unspecific target molecule, the majority of the clones are still unspecific. However, there may also be a population of specific binders in the pool. An additional panning round with increased prepanning can reduce the background. Alternatively, single clone analysis might directly yield specific binders.
Radioimmunoassay is another fast and convenient method to check whether specific target-binding molecules have been enriched in a pool (Hanes et al., 1998). It can be used for the evaluation of both selected pools and individual binders. RNA of a pool or single binder is translated in the presence of radioactively labelled [35S]-methionine. Therefore, the radioactive protein that binds to the surface-immobilized target molecule can be quantified easily. The binding should be performed in the presence and absence of soluble competitor target molecules, and a control for unspecific binding should be included. In the competition assay, the minimal concentration of competitor still leading to half-inhibition of the maximal binding signal is a crude measure for the affinity. Therefore, RIA facilitates the ranking of the affinities of different clones isolated after affinity maturation.
Solutions and Hardware
The general handling is the same as for the in vitro translation. However, the radioactive material must be handled with the appropriate precautions. Do the radioactive work in a designated area of your laboratory. We recommend using filter tips and wearing gloves during the experiment. We recommend doing the RIA in duplicate.
Scintillation counter, ELISA plate shaker
Steps
RNA Preparation
-
- The analysis of whole pools of potential binders can be performed similarly to a normal ribosomedisplay round. A PCR reaction is performed on the selected pool but with primers introducing a 3' stop codon and the standard ribosome display 5' end (including T7 promotor and ribosome-binding site). From this PCR product, RNA is produced as described in Section III,D.
- Alternatively, if you wish to analyze single binders, digest the PCR product from step 1a with the appropriate restriction enzymes and ligate into the ribosome-display vector (RDV). After transformation, isolate plasmids from single colonies. The transcription can be performed directly from the plasmid, which should be present at a concentration of at least 100ng/µl.
- Prepare a Maxisorp plate with immobilized target molecules as described in Section III,E.
- Perform in vitro translations as described in Section III,F with the following modifications: instead of cold methionine use 2 µl of [35S]-methionine (0.3 µM, 50µCi/ml final concentration) per 110-µl reaction and perform translation for 30-45 min at 37°C.
- Stop the translation with 440µl WBT and centrifuge the samples for 5 min at 14,000rpm, 4°C.
- Aliquot the supernatant and dilute the samples with 4% milk in WBT to a final concentration of 1% milk. For inhibition studies, different concentrations of competitor should be added to the different aliquots and the mixture should be equilibrated for 1 h at room temperature prior to step 6.
- Split the samples to at least duplicates and apply them to the blocked plate. Let the protein bind to the target molecule by shaking slowly for maximally 30 min.
- Wash the plate rigorously 5-10 times with WBT.
- Elute bound protein with 100µl of a 0.1M solution of triethylamine (10min at room temperature).
- Transfer the eluates into scintillation tubes containing 5 ml scintillation solution "OptiPhase2."
- Quantify the radioactivity in a scintillation counter.
IV. COMMENTS
This section states some observations we have made during the years of performing ribosome display and gives a summary of how ribosome display can be used for directed evolution experiments.
Naïve libraries, in our hands synthetic antibody libraries or designed repeat protein libraries, are a difficult challenge for selection experiments. The task is to select the specific binders, which are few in numbers, out of a very large number of nonbinders. The outcome of such experiments is not only dependent on the presence of high-affinity binders in the library, but also on the behavior, or "stickiness," of the rest of the library population. In other words, selection must be directed toward specific binding, in contrast to nonspecific binding. This can be achieved with experimental tricks, such as introducing a prepanning step, or by trying to reduce the stickiness of the library population. For selection with naive scFv libraries, it has turned out that six selection rounds were necessary to obtain specific binders (Hanes et al., 2000b). The designed ankyrin repeat protein libraries routinely yield binders after only three to four rounds (Binz et al., 2004). We suspect this might be due to the fact that these repeat proteins, which are extremely well expressed in E. coli, fold well, and are very stable, are also displayed better in vitro than scFv fragments, which are intrinsically somewhat more aggregation prone.
In vitro evolution, the alternation of diversification and selection, is a powerful strategy used to improve proteins. Ribosome display is an ideal platform to perform such experiments. It allows very fast selection cycles and the PCR step is ideal to generate diversity in between the selection rounds. This diversification of the selected pools by random mutations increases the sampled sequence space. Error-prone PCR, e.g., using high Mn2+ concentrations (Leung et al., 1989), imbalanced dNTP concentrations (Cadwell and Joyce, 1994), or nucleotide analogues (Zaccolo and Gherardi, 1999; Zaccolo et al., 1996), is one strategy often applied to achieve diversification. Another powerful strategy to create diversity is DNA shuffling on the selected pools in between the rounds (Stemmer, 1994).
When compared to a selection experiment from a naïve library, the challenge in affinity maturation is different. The applied library will usually be created from a single clone or a pool of clones, which are already good binders. Since in affinity maturation we do not wish to select for binding as such but for better binding, we need an adjustable selection pressure (Jermutus et al., 2001). In principle, two strategies can be used to select tight binders out of a pool of binders.
The second very successful strategy for affinity maturation is off-rate selection. The assumption fundamental to off-rate selection is that the on-rate of most protein-protein interactions is in the range of 105-106 M-1s-1 (Wodak and Janin, 2002) and proteinligand interactions in the range of 106-107 M-1s-1, such that the affinity is largely governed by the off-rate. By first incubating the ribosome-displayed polypeptide with the biotinylated target molecule (typically for 1 h) followed by the addition of a large excess of nonbiotinylated target (1000-fold excess), the selection pressure is governed by the dissociation of the binders from the biotinylated target. While tight binders will remain bound to the biotinylated target, others will dissociate and then rebind to the excess of unbiotinylated target. The incubation time equals the selection pressure and should be adjusted to the expected offrate. If the recovery is poor, it is often useful to include a nonselective round to enrich the binders.
The selection strategies for molecular properties other than high-affinity binding were summarised in Section I. A library can be evolved for these properties, just as it can be for affinity. It exceeds the scope of this chapter to discuss each strategy in detail, and there are many more possibilities that have not been explored experimentally.
Acknowledgment
The authors thank all former and present members of the Pliickthun laboratory involved in ribosome display who helped in developing the present protocol.
References
Amstutz, P., Forrer, P., Zahnd, C., and P16ckthun, A. (2001). In vitro display technologies: Novel developments and applications. Curr. Opin. Biotechnol. 12, 400-405.
Amstutz, P., Pelletier, J. N., Guggisberg, A., Jermutus, L., Cesaro- Tadic, S., Zahnd, C., and Pltickthun, A. (2002). In vitro selection for catalytic activity with ribosome display. J. Am. Chem. Soc. 124, 9396-9403.
Binz, H. K., Amstutz, P., Kohl, A., Stumpp, M. T., Briand, C., Forrer, P., Grtitter, M. G., and Pltickthun, A. (2004). High-affinity binders selected from designed ankyrin repeat protein libraries. Nature Biotechnol. 22, 575-582.
Chen, H. Z., and Zubay, G. (1983). Prokaryotic coupled transcription- translation. Methods Enzymol. 101, 674-690.
Hanes, J., Jermutus, L., and Pltickthun, A. (2000a). Selecting and evolving functional proteins in vitro by ribosome display. Methods Enzymol. 328, 404-430.
Hanes, J., Jermutus, L., Weber-Bornhauser, S., Bosshard, H. R., and Pliickthun, A. (1998). Ribosome display efficiently selects and evolves high-affinity antibodies in vitro from immune libraries. Proc. Natl Acad. Sci. USA 95, 14130-14135.
Hanes, J., and Plfickthun, A. (1997). In vitro selection and evolution of functional proteins by using ribosome display. Proc. Natl Acad. Sci. USA 94, 4937-4942.
Hanes, J., Schaffitzel, C., Knappik, A., and Plfickthun, A. (2000b). Picomolar affinity antibodies from a fully synthetic naive library selected and evolved by ribosome display. Nature Biotechnol. 18, 1287-1292.
He, M., and Taussig, M. J. (1997). Antibody-ribosome-mRNA (ARM) complexes as efficient selection particles for in vitro display and evolution of antibody combining sites. Nucleic Acids Res. 25, 5132-5134.
Jermutus, L., Honegger, A., Schwesinger, E, Hanes, J., and Pliickthun, A. (2001). Tailoring in vitro evolution for protein affinity or stability. Proc. Natl Acad. Sci. USA 98, 75-80.
Kushner, S. R. (2002). mRNA decay in Escherichia coli comes of age. J. Bacteriol. 184, 4658-4665.
Laminet, A. A., and Plfickthun, A. (1989). The precursor of beta-lactamase: Purification, properties and folding kinetics. EMBO J. 8, 1469-1477.
Lesley, S. A. (1995). Preparation and use of E. coli S-30 extracts. Methods Mol. Biol. 37, 265-278.
Leung, D. W., Chen, E., and Goeddel, D. V. (1989). A method for random mutagenesis of a defined DNA segment using a modified polymerase chain reaction. Technique 1, 11-15.
Matsuura, T., and Plfickthun, A. (2003). Selection based on the folding properties of proteins with ribosome display. FEBS Lett. 539, 24-28.
Mattheakis, L. C., Bhatt, R. R., and Dower, W. J. (1994). An in vitro polysome display system for identifying ligands from very large peptide libraries. Proc. Natl Acad. Sci. USA 91, 9022-9026.
Schatz, P. J. (1993). Use of peptide libraries to map the substrate specificity of a peptide-modifying enzyme: A 13 residue consensus peptide specifies biotinylation in Escherichia coli. Biotechnology (New York) 11, 1138-1143.
Smith, G. P. (1985). Filamentous fusion phage: Novel expression vectors that display cloned antigens on the virion surface. Science 228, 1315-1317.
Stemmer, W. P. (1994). Rapid evolution of a protein in vitro by DNA shuffling. Nature 370, 389-391.
Takahashi, E, Ebihara, T., Mie, M., Yanagida, Y., Endo, Y., Kobatake, E., and Aizawa, M. (2002). Ribosome display for selection of active dihydrofolate reductase mutants using immobilized methotrexate on agarose beads. FEBS Lett. 514, 106-110.
Wade, H. E., and Robinson, H. K. (1966). Magnesium ion-independent ribonucleic acid depolymerases in bacteria. Biochem. J. 101, 467- 479.
Wodak, S. J., and Janin, J. (2002). Structural basis of macromolecular recognition. Adv. Prot. Chem. 61, 9-73.
Zaccolo, M., and Gherardi, E. (1999). The effect of high-frequency random mutagenesis on in vitro protein evolution: A study on TEM-1 β-1actamase. J. Mol. Biol. 285, 775-783.
Zaccolo, M., Williams, D. M., Brown, D. M., and Gherardi, E. (1996). An approach to random mutagenesis of DNA using mixtures of triphosphate derivatives of nucleoside analogues. J. Mol. Biol. 255, 589-603.
Zubay, G. (1973). In vitro synthesis of protein in microbial systems. Annu. Rev. Genet. 7, 267-287.
Support our developers




