Familial Hypercholesterolemia
Brown and Goldstein studied LDL metabolism in cells from patients with a common metabolic inherited disorder called familial hypercholesterolemia (FH). People homozygous for this mutation have a 6- to l0-fold elevation of LDL levels, are born with detectable atherosclerosis, and usually do not survive childhood without a myocardial infarction. Heterozygotes have two- to fourfold elevations in LDL and suffer from coronary heart disease (CHD) during middle age (85% of FH heterozygotes have a heart attack before the age of 60.) FH is the most common inherited metabolic disorder in humans, with a gene frequency of 1 in 500, i.e., 1 in 500 people is a heterozygote for FH.Brown and Goldstein discovered that FH cells from homozygote donors showed little or no LDL-binding activity. FH cells from heterozygotes possessed about 50% of normal activity. They concluded that mutations in the gene encoding the LDL receptor are the molecular basis for the FH disease. The inability to clear LDL through the normal LDL receptor pathway causes hypercholesterolemia atherosclerosis.
The loss of LDL receptor activity readily explains the inefficient clearance ofLDLand the hypercholesterolemia of FH patients (Fig. 9). In addition to defective catabolism of LDL, there is also LDL overproduction for the following reason. IDL is also cleared through the LDL receptor. Diminished LDL receptor activity leads to prolonged circulation of IDL, giving it a greater opportunity to be converted to LDL. IDL is at an important branch point in lipoprotein metabolism; it can be directly cleared from the circulation or it can be further processed to become LDL. The LDL receptor can also regulate the secretion of VLDL. It promotes reuptake of newly secreted VLDL. Also, within the secretory pathway, the LDL receptor promotes the degradation of newly synthesized apo-B100.
The principal regulatory site in the cholesterol biosynthetic pathway is the step catalyzed by the enzyme 3-hydroxy-3-methylglutaryl-CoA reductase (HMG-CoA reductase; Fig. 10). Regulation of HMG-CoA reductase by LDL cholesterol occurs principally through the regulation of the level of its mRNA. In analogy to genes regulated by steroid hormones, nuclear transcription factors bind to DNA sequences upstream from the reductase gene and regulate transcription. In addition to transcriptional regulation, cholesterol and certain other sterols diminish HMG-CoA reductase by a second mechanism; they hasten the degradation of the enzyme. The protein is located at the ER membrane and has several transmembrane domains that are necessary in order for cholesterol to stimulate the degradation of the protein.
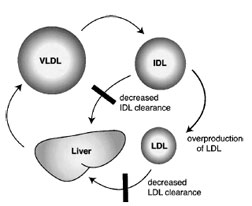 |
| Figure 9 Kinetic mechanism for LDL overproduction in familial hypercholesterolemia. VLDL catabolism gives rise to IDL. IDL has two competing fates. It can be cleared by the liver or continue to be processed to become LDL. In the absence of the LDL receptor, IDL clearance is sluggish, thus a large proportion of IDL is converted to LDL. |
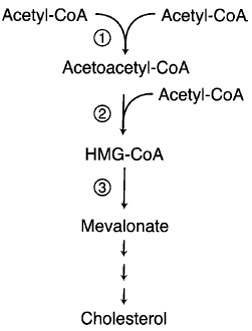 |
| Figure 10 Regulated steps in cholesterol synthesis pathway. Step 1 is catalyzed by cytosolic acetoacetyl-CoA synthase. Steps 2 and 3 are catalyzed by HMG-CoA synthase and HMGCoA reductase, respectively. The later two enzymes are transcriptionally regulated by SREBP. Cholesterol feeds back on its own synthesis by decreasing the abundance of enzymes 2 and 3. HMG-CoA reductase is the target of widely used cholesterollowering drugs known as “statins.” Between mevalonate and cholesterol are more than 30 steps and branch points to nonsteroidal isoprenoid molecules. |
Support our developers




