Tangier Disease and Familial Hypoalphalipoproteinemia
Tangier disease is a rare recessive disorder in which patients have almost no HDL. Cholesterol ester accumulates in macrophages and macrophage-rich tissues like spleen and liver. Familial hypoalphalipoproteinemia (FHA) is a very common dominant disorder in which people have low HDL (typically < 30 mg/dl) and suffer from premature heart disease even without an elevation in LDL.Approximately 40% of patients with premature coronary heart disease have low HDL, making it the most common lipid disorder of heart disease patients.
Tangier disease and FHA are caused by mutations in the same gene, ABCA1. Tangier disease patients are homozygous (or inherit two different mutant alleles). FHA patients are heterozygous for mutations at the ABCA1 locus. ABCA1 mutations lower HDL because they prevent phospholipid and/or cholesterol from effluxing and becoming associated with apo-A1 and apo-A2. In the absence of sufficient lipid, apo-A1 is rapidly cleared by the kidneys.
Why do mutations in ABCA1 lead to premature heart disease? ABCA1 fulfills a rate-limiting step in the pathway by which cells get rid of cholesterol, and thus might be a critical protector against cholesterol overload. With the exception of hepatocytes, cells are unable to catabolize large quantities of cholesterol and must therefore protect themselves from cholesterol overload by expelling cholesterol to an appropriate extracellular carrier. It is interesting that ABCA1 is especially abundant in macrophages; macrophages can become engorged with cholesterol esters and form foam cells in the arterial wall. A mutation in ABCA1 might therefore predispose an individual to atherosclerosis by impeding cholesterol efflux.
Do HDL levels correlate with the rate of reverse cholesterol transport? Studies of the expression of the SR-B1 receptor provide an answer to this question. When HDL binds to SR-B1 at the plasma membrane of the hepatocyte, it unloads its cholesterol ester cargo. The lipid-depleted apo-A1 then is cleared from the circulation, in part by the kidney. High levels of SR-B1 therefore lead to low HDL levels. However, under these circumstances, the transport of cholesterol to the liver is enhanced. When the diet is shifted from saturated to polyunsaturated fat, HDL levels typically drop. This drop is attributable to a rise in SR-B1 expression in the liver. In short, a decrease in HDL can be caused by imparied reverse cholesterol transport (extrahepatic; Tangier disease) or by increased reverse cholesterol transport (liver; polyunsaturated fat).
HDL metabolism is intimately connected with triglyceride metabolism. Clinicians know this because patients with hypertriglyceridemia almost invariably have low HDL levels. The cholesterol ester transfer protein (CETP)-catalyzed transfer of cholesterol ester from HDL to VLDL occurs with reciprocal transfer of triglyceride to HDL (Fig. 13). Lipoprotein lipase and hepatic lipase (another cell-surface-bound lipase) catalyze the hydrolysis of HDL triglyceride, reducing the size of the HDL particles. Hepatic lipase can also hydrolyze HDL phospholipids. Smaller HDL particles (HDL3) are removed from the circulation more quickly than larger HDL (HDL2).
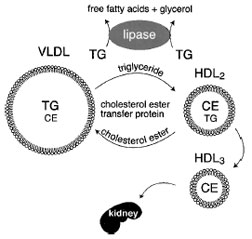 |
| Figure 13 Inverse relationship between plasma triglyceride and HDL cholesterol levels. A higher level of VLDL correlates with lower HDL levels. Two processes simultaneously remove triglycerides from VLDL particles. First, lipoprotein lipase hydrolyzes the triglycerides to free fatty acids and glycerol. Second, cholesterol ester transfer protein (CETP) in the bloodstream catalyzes the exchange of triglyceride and cholesterol ester between VLDL and HDL, respectively. As HDL accumulates triglyceride, it is a substrate for lipoprotein lipase and hepatic lipase. This shrinks the HDL particles, causing them to be cleared by the kidneys. |
The abundance of VLDL relative to HDL significantly influences HDL metabolism, probably due to enhanced exchange of triglyceride into the HDL particles. Individuals with genetically reduced levels of cholesterol ester transfer protein have extremely high HDL levels. Heavy exercise is also associated with increased HDL levels. Exercise increases the expression of muscle lipoprotein lipase. The resulting increase inVLDLlipolysis decreases the amount of triglyceride that can participate in the CETP lipid exchange process. This results in an elevation in HDL.
Support our developers




