Chemical Transformations
The chemistry of carbohydrates involves transformations at both the carbonyl function and the chain hydroxyl groups. The presence of multiple species of simple sugars in aqueous solution gives rise to complex reaction patterns wherein product distribution may well be determined by kinetic rather than thermodynamic considerations.1. Reactions at the Carbonyl Group
The carbonyl function reacts directly with a variety of reagents either as the free aldehyde or keto group or as the corresponding hemiacetal or hemiketal. In early work, Fischer introduced phenylhydrazine, which condenses at the carbonyl carbon and oxidizes the adjacent secondary hydroxyl (aldoses) or primary alcohol (ketoses) to allow a second condensation. The resulting derivatives, phenylosazones, are crystalline and were employed both for identification and determination of stereochemistry. Phenylhydrazine itself is a liver poison and it is generally believed that Fischer suffered from liver damage due to this reagent (no longer employed). A simple example of the utility of this conversion is the fact that D-glucose, D-mannose, and D-fructose all give the same phenylosazone showing that their relative stereochemistry at the remaining asymmetric centers (carbons 3,4,5) is identical (Fig. 13).
Certain transformations may dictate that the carbonyl function remain available for later chemistry while reactions are carried out elsewhere in the chain. This may be achieved by formation of dithioacetals or, in the ring form, by suitable substitution of the anomeric hydroxyl group (see below).
2. Reaction at the Anomeric Hydroxyl Group
The hemiacetal nature of the anomeric hydroxyl group makes it the most reactive of that type. Direct oxidation can be carried out with several reagents, most classically, hypoiodite. This results in rapid oxidation to the aldonolactone and is only exhibited by aldoses. Alternatively, reaction with alcohols results in the formation of full acetals (glycosides). A very large variety of such structures have been made.
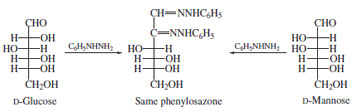 |
| Figure 13 Formation of phenylosazone from D-glucose or D-mannose. The loss of asymmetry at C-2 yields identical products. |
3. Reactions at Secondary Hydroxyls
Substitution reactions at secondary hydroxyls are generally performed either for analysis of structure or to serve a protective function during other reactions. Etherification of the nonanomeric hydroxyls was an important structural tool in the analysis of oligosaccharide and polysaccharide structure. Methyl ethers have been employed for structural determination for more than 75 years. Thus, methyl ether formation in a polysaccharide results in substitution only at free hydroxyls. Subsequent analysis of the methylated derivatives reveals positions previously occupied in glycosidic linkage. Reagents used for this purpose have evolved from dimethylsulfate to the commonly employed method of Hakomori using sodium hydride and dimethylsulfoxide.
Another frequently used ether substituent is the benzyl group, which is stable under a variety of reaction conditions but can be removed by catalytic hydrogenation. This is employed mainly during synthetic schemes where protection of specific hydroxyls is required. Currently, ether formation is used primarily as an adjunct to mass spectrometric analysis.
The secondary hydroxyl groups can also be esterified. Acetyl substitution via acetic anhydride is often used, especially as a protective group. Tosyl substitution using toluenesulfonyl chloride is often employed since the properties of that ester (good leaving group) allow for SN2 displacement resulting in inversion of configuration. Acetate esters are readily removed under basic conditions (methoxide ion) when glycosides remain intact.
4. Reactions at the Hydroxymethyl Group
The exocyclic nature of this group makes it the second most reactiveof thehydroxyl functions.Tritylationis often employed to transiently block C-6 (ready removal under mild acid conditions). This is also the site of enzymatically catalyzed phosphorylation of glucose and other sugars, the first step in their metabolic utilization. Oxidation to a carboxylic acid is common. The resulting uronic acid is, for the common sugars, widely distributed in natural polysaccharides in both plants and animals. It is of interest that L-iduronic acid (the 5-epimer of D-glucuronic acid) and L-guturonic acid (the 5-epimer of D-mannuronic acid) are formed in nature after the respective precursor uronic acid has been incorporated into polymeric linkage.
5. Hydroxyl Pairs
The proximity and defined stereochemistry of hydroxyl groups in the ring structures of saccharides allows for a number of selective reactions. These include formation of acetals or ketals and of a group of interactions restricted to hydroxyls on adjacent carbons.
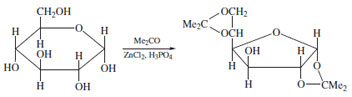 |
| Figure 14 1,2 5,6-diisopropylidene D-glucofuranose. Formed by reaction of D-glucose with acetone in the presence of a suitable catalyst. The furanose product dominates due to kinetic control of the reaction. |
Adjacent hydroxyls that are cis can undergo several different reactions. Included in this group is complexation with borate to form a transient cyclic adduct (Fig. 15). This alters chemical reactivity and electrophoretic properties of the reactive sugar and has several applications.
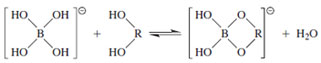 |
| Figure 15 Borate ester formation. cis-Hydroxyls are preferred. The extent of reaction was originally monitored by following the change in conductivity of borate solutions on the addition of saccharide. |
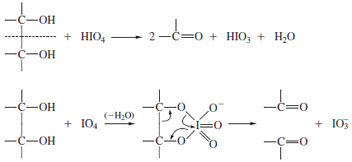 |
| Figure 16 Action of periodate on vicinal diols. The proposed cyclic intermediate suggests that rigid structures with transhydroxyls will react poorly, a prediction confirmed experimentally. |
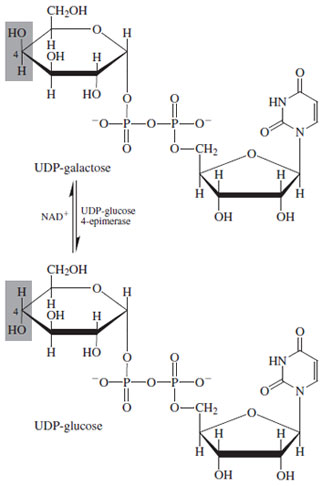 |
| Figure 17 Biosynthesis of D-galactose by epimerization of uridiine diphosphoglucose. |
Support our developers




