The Ribosome
The bacterial ribosome has been the subject of intense study for several decades. Although the general mechanisms of protein synthesis (as outlined earlier) are reasonably well understood, only recently have structures emerged which make a molecular description of ribosome function appear possible. Because of the high degree of functional and sequence conservation between bacterial and eukaryotic components of the ribosome, structural information obtained using bacterial ribosomes is expected to contribute to a universal understanding of ribosomal architecture.A. Composition of the Ribosome
The E. coli ribosome contains 3 ribosomal RNA (rRNA) molecules and about 50 proteins, divided between two unequal subunits. (The number of proteins is not entirely certain, as some proteins are loosely associated with the ribosome but may not be integral to its function.) The small subunit sediments as a 30S particle, which has a single 16S rRNA molecule and 21 proteins, S1–S21 (S in this case indicates a small subunit protein). The large subunit has two rRNAs (5S and 23S) and at least 34 proteins, with L-prefixes. In the cell, ribosomal subunits spontaneously assemble from their protein and rRNA components and are brought together as a functional complex with mRNA and tRNA at the initiation step of translation. Individual subunits and functional ribosomes can also be reconstituted in vitro under the right salt and temperature conditions. The determination of these conditions has greatly facilitated structural studies of the ribosome. For example, subunits can be reconstituted in the absence of one or more proteins and then assayed for structural integrity and function. Such experiments determined which proteins are essential, either because they are needed for subsequent binding of other proteins or because they are necessary for ribosomal function.
B. Sequence Conservation of Ribosomal Components
Nucleotide sequences are now known for rRNAs from hundreds of organisms. Comparisons of these sequences allowed predictions of the secondary structures of these RNAs (which regions are single stranded and which base pair to form helices). Interestingly, the rRNA secondary structures are evolutionarily conserved, while the individual nucleotides that make up these structures often are not. Only small stretches of rRNA are identical in the majority of organisms, and these are therefore predicted to be critical for ribosomal function.
More sequence conservation is observed for ribosomal proteins than for rRNA. Structural similarities are significant enough that cross-species RNA interactions were shown possible for proteins L1, L11, and S15. At a minimum, then, the RNA-binding features of these proteins are conserved.
C. Structural Studies of the Ribosome
The ribosome’s size challenged structural studies since its first observation in electron micrographs nearly 50 years ago. With a molecular mass of 2500 kD and dimensions about 250 Å on a side, the 70S ribosome is small for electron microscopy but immense for structural studies (such as NMR and X-ray crystallography) typically applied to single molecules.
The size and complexity of the ribosome have resulted in the development and application of many different experimental approaches to probe the arrangement of proteins and rRNA. Electron microscopy hinted at the overall shape of individual subunits and the assembled ribosome. The delineation of these shapes has changed little even with current high-resolution structures. The generation of antibodies against ribosomal proteins, rRNA termini, and modified bases allowed identification of their respective locations on the ribosome surface by means of immunoelectron microscopy. Specific sites of protein–rRNA interactions were determined by chemical modification experiments, which generate a “footprint” of protein binding according to the differential reactivity of rRNA nucleotides in the presence and absence of the protein. Similar modification experiments also confirmed the secondary structure predictions made for rRNAs, using enzymes and chemicals that react selectively with bases in either singlestranded or helical regions.
More detailed information was obtained about the orientation of proteins and rRNA, using reagents that generate covalent cross-links between ribosomal neighbors. Protein–protein, protein–RNA, and RNA–RNA crosslinks provided valuable constraints for model-building studies by limiting the distance that these components could be placed from one another according to the length of the cross-linker. A related approach has been the use of reagents that cleave rRNA. The cleavage patterns reveal parts of the rRNA that are either buried or exposed.
Neutron diffraction of ribosomal proteins has been used to estimate the arrangement of ribosomal components. By reconstituting pairs of deuterated proteins into the 30S subunit, distances between the protein pairs were generated to produce a model of protein arrangement in the small subunit. Using similar methods several pairwise distances have been generated for the large subunit as well but, with over twice as many proteins, the task is significantly more difficult.
Together the accumulated biochemical and structural data have generated several models of ribosome structure. Several high-resolution structures of individual ribosomal proteins or fragments of rRNAbound to proteins have also been determined. However, the piecewise reconstruction of structural data seemed unlikely to provide an adequate picture of the ribosome at the molecular level. Recently, however, structures of isolated subunits and the assembled ribosome have been determined at a resolution that is already impressive and will continue to improve.
D. Recent High-Resolution Structures
Neither electron microscopy nor X-ray crystallography seemed suited to provide a high-resolution structure of the ribosome, which was thought to be too small for the first and too large for the second method. Yet both have been pushed to the limit to provide significant new structures that are already explaining mechanistic features of protein synthesis.
Cryoelectron microscopy (cryo-EM) has produced remarkable images of the entire ribosome as a result of two major advances in methodology—one experimental, one computational. Samples for cryo-EM (in this case of the 70S E. coli ribosome) are frozen quickly, encased in vitreous ice which lacks the crystals that can distort macromolecular structure. Following electron microscopy of these samples, a computer program digitally averages and aligns tens of thousands of images from single ribosomes at different angles. The resulting reconstruction produces a three-dimensional image at much higher resolution than any of the individual two-dimensional micrographs.
One of the first features observed by cryo-EM was the amount of visible deep clefts and “holes.” Intersubunit contacts are limited to a discrete number of “bridges” between large and small subunits. These now appear to be composed primarily of RNA. The relatively small intersubunit surface is consistent with the need for subunits to dissociate following protein synthesis to accept a new mRNA molecule.
![Bacterial ribosome structure at 7.8 Å. Architectural features of Thermus thermophilus 70S ribosomes are labeled, as identified by X-ray crystallography. The 30S subunit is the front, darker portion, and consists of the head (H) connected to the platform (P) and body (B). Other features of the 30S subunit are the neck (N), spur (SP), shoulder (S), and contacts between the head and platform (a and b). The 50S subunit includes the protein L1 stalk, central protuberance (CP), and L7/L12 region. [Reprinted with permission from Cate, J. A., Yusupov, M. M., Yusupova, G. Zh., Earnest, T. N., and Noller, H. F. (1999). “X-ray crystal structures of 70S ribosome functional complexes.” Science 285, 2095-2104. © 1999 American Association for the Advancement of Science.]](images/12-11.jpg) |
| Figure 11 Bacterial ribosome structure at 7.8 Å. Architectural features of Thermus thermophilus 70S ribosomes are labeled, as identified by X-ray crystallography. The 30S subunit is the front, darker portion, and consists of the head (H) connected to the platform (P) and body (B). Other features of the 30S subunit are the neck (N), spur (SP), shoulder (S), and contacts between the head and platform (a and b). The 50S subunit includes the protein L1 stalk, central protuberance (CP), and L7/L12 region. [Reprinted with permission from Cate, J. A., Yusupov, M. M., Yusupova, G. Zh., Earnest, T. N., and Noller, H. F. (1999). “X-ray crystal structures of 70S ribosome functional complexes.” Science 285, 2095-2104. © 1999 American Association for the Advancement of Science.] |
In addition to unoccupied 70S ribosomes, cryo-EM reconstructions are currently available for ribosomes containing various ligands. For example, tRNAs have been observed in each of the three ribosomal binding sites (A, P, and E), and ribosome-bound EF-Tu has also been visualized. The variety of structures available at up to 11 Å resolution points out an advantage of cryo-EM over X-ray crystallography for ribosomal structure determination. Because sample preparation is rapid compared to growth of crystals for X-ray diffraction studies, the many functional states of the ribosome can be more readily probed by EM to assemble a gallery of structures.
Even more surprising than the impressive cryo-EM structures have been recent leaps in resolution obtained by X-ray crystallography of ribosomes (Fig. 11). Although researchers have been able to generate diffracting crystals of ribosomes for years, only recently has the diffraction been of sufficient quality to identify clear regions of electron density corresponding to rRNA and protein. Here the lower-resolution cryo-EM structures have helped, in that they enabled crystallographers to position and orient the ribosomal particle within the crystal unit. Once the ribosome is properly oriented in the crystal, the heavy atom clusters used to determine phase angles can be located. Together these advancements provided remarkable images at moderate resolution of individual ribosomal subunits and the complete particle with mRNA and tRNAs bound. Atomic resolution structures will soon be forthcoming.
The structures now available at 5–8 Å resolution allow differentiation of protein α-helices from double-helical regions of rRNA based on their structural characteristics; some protein β-sheets can also be seen. The structures of several individual ribosomal proteins have been solved by X-ray crystallography or nuclear magnetic resonance spectroscopy, and the X-ray data on the whole 50S particle have been correlated with the previously determined structures of individual proteins. As expected, the accumulated data from IEM, cross-linking, and neutron diffraction helped to position these protein structures within the ribosome electron density and will continue to direct placement of proteins and specific regions of rRNA.
Although the locations of many proteins and thousands of nucleotides must still be teased out of the electron density maps, structural features of specific regions of the ribosome have already provided mechanistic clues. A more detailed understanding of the mechanism of protein synthesis will emerge from these structural studies.
E. Mechanistic Clues from Structure
On the interface side of the 30S subunit (where it interacts with the 50S subunit), a large region of rRNA is visible which lacks any protein density. This observation supports the theory that a primitive ribosome was composed only of RNA. Localization of the proteins of known structure revealed that S5 makes close contacts with S4; these were previously known to be near each other based on cross-linking and neutron diffraction data. Mutations in S4 and S5 produce error-prone ribosomes, and in fact these mutations are mapped to the area of contact between the two proteins. It seems likely, therefore, that inaccuracy results from a disruption of this protein–protein interaction.
A major feature observed on the 50S subunit structure is an apparent tunnel through the center of the subunit, through which the newly synthesized polypeptide is proposed to exit the ribosome. The tunnel seems to be wide enough to accommodate the polypeptide, and extends from the peptidyl transferase center to A-site on the back (solvent side) of the 50S subunit.
The crystal structure of the Thermus thermophilus 70S ribosome was solved with RNA substrates in the A, P, and E-sites of the ribosome and with bound mRNA. Interestingly, a partial tRNA mimic (an isolated anticodon stemloop hairpin) bound in the P-site, held tightly in place by six contact points with the ribosome. These contacts likely stabilize the codon–anticodon interaction, properly orient the anticodon stem in the P-site, and may help position the A-site codon of themRNA. The tRNAmimic bound in the A-site is not held tightly, but sits in a large cavity of electron density with only minimal contact points. This makes sense mechanistically, as the A-site serves to discriminate between cognate and noncognate tRNAs, while the P-site must grip the peptidyl–tRNA to prevent dissociation from the ribosome.
F. Dynamics of Translation
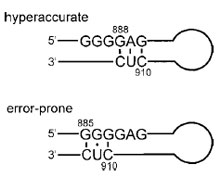 |
| Figure 12 The 16S triplet switch. Genetic studies revealed that the 885–912 regions of 16S rRNA alternate between base-pairing conformations, as shown; both are required for cell viability. Mutations that favor the 888 conformation produce ribosomes that are hyper- accurate in protein synthesis, while substitutions that stabilize the 885 conformation lead to error-prone phenotypes. It is likely that this switch is involved in regulating tRNA selection and translocation. |
For example, the 16S nucleotides 885–890 and 910– 912 can base pair in two different conformations (Fig. 12). The 910–912 CUC can form canonical Watson–Crick base pairs with the GAG of 888–890 or can pair with the GGG of 885–887 to give a central G–U wobble pair. Substitutions demonstrated that both pairings are essential for biological activity, suggesting that 16S rRNA switches between two conformations at different steps in protein synthesis. Furthermore, mutations that favored the “888 conformation” led to restrictive (hyper-accurate) ribosomal phenotypes, while those that stabilized the “885 pairing” produced error-prone phenotypes. Structurally, this triplet-switch-sequence has been located in the 70S crystal structure near the long 16S helix that is part of the decoding region of the small subunit. The switch helix is certainly involved in tRNA selection, and may also play a role in translocation, as suggested by the observation that mutations that favored the 888 conformation were hypersensitive to spectinomycin, an antibiotic known to inhibit EF-G-dependent translocation.
Support our developers




