Cultured for Neuronal PC12 Ce lls: A Model Function, Differentiation, and Survival
Since its initial description and characterization in 1976 (Greene and Tischler, 1976), the rat pheochromocytoma PC12 cell line has become a commonly employed model system for studies of neuronal development and function. In particular, PC12 cells have been a convenient alternative to cultured neurons for studying the trophic and differentiative actions of nerve growth factor (NGF; reviewed by Levi-Montalcini and Angeletti, 1968; Levi and Alem& 1991). When cultured in serum-containing medium, PC12 cells adopt a round and phase-bright morphology and proliferate to high density. Under these conditions, PC12 cells display many of the properties associated with immature adrenal chromaffin cells and sympathicoblasts. When challenged with physiological levels of NGF, these cells cease division, become electrically excitable, extend long branching neurites, and gradually acquire many characteristics of mature sympathetic neurons. Under serum-free conditions, NGF promotes not only neuronal differentiation of PC12 cells, but also their survival (Greene, 1978; Rukenstein et al., 1991).
II. MATERIALS AND INSTRUMENTATION
Rosewell Park Memorial Institute 1640 (RPMI 1640) medium is purchased from Invitrogen (Carlsbad, CA; Cat. No. 23400062) in powder form. Donor horse serum (Cat. No. 12-44977P), fetal bovine serum (Cat. No. 12-10377P), and penicillin/streptomycin (Cat. No. 59-60277P) are obtained from JRH Biosciences (Lenexa, KA). It is recommended that sera be prescreened for their capacities to promote PC12 cell growth and maintenance The horse serum should be heat inactivated in a 56°C water bath for 30min before use. Tissue culture plasticwares are obtained from Falcon, Becton Dickson and Company (Lincoln Park, NJ). Freezing vials are purchased from Nunc, Denmark (Cat. No. 377267). Millipak-60 filters (0.22 µm, Cat. No. MPGL06SH2) are from Millipore (Bedford, MA). Filter units (0.45 µm, Cat. No. 245-0045) are obtained from Nalgene Company (Rochester, NY). Ethylhexadecyldimethylammonium bromide (Cat. No. 117 9712) is purchased from Eastman Kodak Company (Rochester, NY).
Rat tail collagen is prepared in 0.1% acetic acid as described previously (Greene et al., 1991) from the tendons of rat tails [see Fig. 14.2 of Kleitman et al. (1991) for a photographic illustration of the procedure for exposing and removing rat tail tendons]. Each large tail furnishes approximately 200ml of stock collagen. Aliquots of collagen stock are stored at-20°C. Once thawed, the stock can be stored for up to several months at 4°C. Sterile technique should be employed throughout the preparation.
III. PROCEDURES
A. Routine Tissue Culture Techniques
Solutions
- Complete growth medium: Prepare RPMI 1640 medium according to the supplier's protocol in reverse osmosis/Milli-Q or double-distilled water. After the addition of sodium bicarbonate (2g/liter), penicillin (final concentration 25 U/ml), and streptomycin (final concentration 25 µg/ml), sterilize the medium by pressure filtration (driven by 90% air, 10% CO2 mixture) through a Millipak-60 filter unit, dispense into 500-ml autoclaved bottles, and store in the dark at 4°C. The bottles should be dedicated to tissue culture only and should be cleaned by thorough rinsing without soap or detergent. To make up complete growth medium, add 50 ml of heat-inactivated horse serum and 25 ml of fetal bovine serum to 500ml of RPMI 1640 medium. Store complete growth medium at 4°C.
- Medium for freezing of cells: Mix 1 volume of dimethyl sulfoxide (DMSO) with 9 volumes of complete growth medium. This medium should be freshly prepared for immediate use only.
- PC12 cells show optimal adherence to collagencoated culture dishes. Before applying to dishes, freshly dilute the stock collagen solution with autoclaved reverse osmosis/Milli-Q water as noted later. The optimal final dilution of the collagen should be determined empirically by testing various concentrations for their capacities to foster cell attachment and NGF-promoted neurite outgrowth (Greene et al., 1991). At too low a dilution, adhesion to substrate is poor, whereas at too high a concentration, neurite outgrowth is impeded and cells are difficult to dislodge for subculture. For application to plates without the necessity of spreading a thin layer, add the diluted collagen solution (typically a 1:50 dilution of the stock is optimal) to plastic tissue cultureware (10 ml/150-mm dish; 5 ml/100-mm dish; 1 ml/35-mm dish; 0.5 ml/well of 24-well culture plates). Leave dishes uncovered to air dry overnight in a tissue culture hood. Alternatively, for quicker drying, dilute the collagen by a factor of five less and add to cultureware in one-fifth the aforementioned volumes. Spread the collagen evenly over the surface of the culture dish with the use of an Lshaped glass rod. Dry collagen by leaving the plates uncovered for 1-2h in a tissue culture hood. Store collagen-coated dishes at room temperature and use within 1 week after preparation.
- Feed PC 12 cells three times a week with complete growth medium. Remove approximately twothirds of the culture1 medium from each plate and replace with fresh complete growth medium (10 ml for 150-mm dishes; 5 ml for 100-mm dishes; 1.5 ml for 35- mm dishes). The medium should be added gently and from the side of the tissue culture dish. The feeding schedule should be kept rigid for maximum cell viability. Maintain PC12 cells in a 37°C incubator with a water-saturated, 7.5% COs atmosphere.
- Passage PC12 cells (subcultured) when the cultures are 80-90% confluent. Dislodge the cells from the surface of the dish by repeated and forceful discharge of the culture medium directly onto the cells with a disposable glass Pasteur pipette. Forceful trituration of the cell suspension within the Pasteur pipette also decreases cell clumping. Mix the culture medium containing detached PC12 cells with fresh complete medium in a 1:3 or 1:4 ratio. Plate the PC12 cells onto collagen-coated dishes, and increase the passage number of the newly plated PC12 cells by one. As the cell doubling time is 3-4 days, subculture every 7-10 days. To avoid the potential accumulation of variants within the cultures, experiments should be carried out with cells that have undergone no greater than 50 passages.
- Stock cultures of PC12 cells are frozen at high density (>5 × 106 cells/ml; see later for cell counting procedure) as follows. Dislodge cells from tissue culture dish as described earlier, pellet by centrifugation at room temperature for 10min at 500g, and remove the medium. Add the appropriate volume of freezing medium (described earlier) and resuspend the cell pellet. Aliquot into a Nunc freezing vial (1 ml/vial) and transfer to a-80°C freezer for at least 1 day. For high-viability, long-term storage, the vials should be maintained in liquid nitrogen. The vials should not be permitted to warm up during the transfer to liquid nitrogen (e.g., transfer on dry ice).
- Thaw frozen PC12 cell stocks (in freezing vials) rapidly in a 37°C water bath (2-3min). Immediately transfer the cells into 9 volumes of complete growth medium. Pellet the cells by centrifugation at room temperature for 10min at 500g. Discard the supernatant. Resuspend the cell pellet in fresh, complete growth medium and plate cells on collagen-coated dishes.
Solution
Low serum medium: Mix 1 ml of heat-inactivated horse serum per 100 ml of RPMI 1640 medium. Store at 4°C.
Steps
- Dislodge PC12 cells from stock culture dishes and triturate well using a glass Pasteur pipette to break up cell clumps. Then plate cells at low densities (5 × 106 cells per 150-mm dish; 1-2 × 106 cells per 100-mm dish; 2-5 × 105 cells per 35-mm dish; see cell-counting procedure described later) on collagen-coated dishes in medium supplemented with NGF (50-100ng NGF final concentration/ml of medium). Dilute NGF from the stock into the medium just before use. Because NGF binds to glass surfaces, use plasticware. Diluted solutions of NGF are not stable. Although neurite outgrowth is satisfactory in complete growth medium, low serum medium is recommended instead in order to economize on serum as well as to reduce cell clumping. Once plated, the cultures should be maintained in a 37~ incubator with a water-saturated, 7.5% CO2 atmosphere and exchange the medium three times per week as described earlier. Neurite-bearing cells should be noticeable within 1-3 days of NGF treatment, and the number of PC12 cells with neurites should increase progressively with time of NGF exposure. By 7-10 days of treatment, at least 90% of the cells should generate neurites.
- To determine the proportions of neurite-bearing PC12 cells after NGF treatment, observe cultures with a phase-contrast microscope under high magnification (e.g., 200×). Within random fields, score the proportions of cells that possess at least one neurite greater than 20µm (about two cell body diameters) in length. Continue counting until the total number of cells assessed exceeds 100. For consistent results, count only discernible and/or single cells, but not cell clumps. To determine mean neurite lengths and rates of neurite elongation, observe cultures using an eyepiece equipped with a calibrated micrometer. The latter is used to measure the entire length of randomly chosen neurites. At least 20-25 neurites are measured per culture.
- Neurite regeneration experiments are carried out with NGF-pretreated PC12 cell cultures. This permits study of rapid neurite growth as well as a quantitative bioassav for NGE Treat the cells first with NGF for 7-10 days as described in step 1. Then rinse the cultures five times with medium (without NGF) while the cells are still attached to the substrate. Mechanically dislodge the cells from the dish by trituration through a Pasteur pipette and wash them an additional five times in medium (without NGF) by repeated centrifugation at 500g for 10min at room temperature. Plate the washed cells at low density (about 105 cells/35-mm dish) in medium (complete or low serum) in the presence or absence of NGF (see step 1). Examine the cultures 24h later and score for percentage of neurite-bearing cells or cell clumps. Because NGFtreated PC12 cells tend to aggregate, it is often necessary to score clumps rather than single cells. The ability of NGF to induce neurite regeneration from PC12 cells is determined by subtracting the number of neurite-bearing cells in culture medium without NGF from the number of neurite-bearing cells in NGFcontaining culture medium. For well-washed cultures, 80-100% of the cells or cell clumps should regenerate neurites with NGF, whereas no more than 10-20% should regenerate neurites in the absence of NGE The regeneration protocol can be used as a quantitative bioassay for NGF (Greene et al., 1987).
Solutions
- Nuclei counting solution stock (Soto and Sonnenschein, 1985): Dissolve 5g ethylhexadecyldimethylammonium bromide and 0.165g NaCl in 80ml of reverse osmosis/Milli-Q water. Add 2.8 ml glacial acetic acid and I drop bromphenol blue. Bring final volume to 100 ml and filter through a 0.45-µm filter unit. Store the solution at room temperature.
- Working nuclei counting solution (Soto and Sonnenschein, 1985): Mix phosphate-buffered saline (10ml), 10% Triton X-100 (5 ml), 1M MgCl2 (200 µl), and nuclei- counting solution stock (10 ml) with enough reverse osmosis/Milli-Q water so that the final volume is 100ml. Pass through a 0.45µm filter unit. Store the working nuclei-counting solution at room temperature.
Steps
- To determine the numbers of PC12 cells suspended in culture or other medium, pellet the cells by centrifugation, aspirate to remove the medium, and resuspend the cells in a known volume of the working nuclei-counting solution. This solution provides a homogeneous suspension of intact nuclei, which are quantified using a hemocytometer. To count cells attached to a substrate, remove the medium and replace with a known volume of working nucleuscounting solution. Resuspend nuclei by trituration and quantify with a hemocytometer.
- Wash PC12 cells (either naive cells growing with serum or neuronally differentiated cells growing with serum and NGF) with serum-free RPMI medium five times while still attached to culture dishes and then, after detachment by trituration, wash another five times in serum-free RPMI medium by centrifugation/ resuspension. Resuspend the cells in RPMI 1640 medium with or without NGF or other potential trophic agents. Plate the cells in collagen-coated, 24- well culture plates in 0.5ml of medium (0.5-2 × 105 cells/well). Exchange the serum-free culture medium three times per week. Carry out cell counts by removing the medium, adding working nuclei-counting solution, and counting intact nuclei. Typically, without trophic substances such as NGF, 50% of the cell die by 24h of serum deprivation and 90% by 3-4 days.
Solutions
OptiMEM-I and Lipofectamine2000: Both reagents are purchased from Invitrogen. OptiMEM-I is used for transfection without antibiotics.
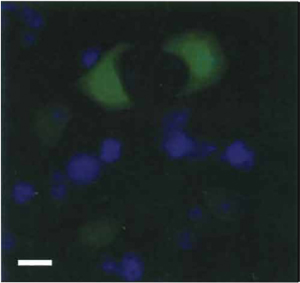 |
| FIGURE 1 PC12 cells expressing green fluorescent protein (GFP). Photomicrograph of non-NGF-treated (naive) PC12 cells transfected with plasmid expressing GFP (green), followed by DAPI staining (blue). Bar: 50µm. |
- As noted in Section I, an advantage of PC12 cells is that they can be subjected to genetic manipulation. A relatively high transient transfection efficiency (at least 20-30%) of PC12 cells is attainable by the use of lipid-based transfection reagents (Fig. 1). Following transfection, stable lines of PC12 transfectants can also be obtained by applying the appropriate selection pressure (e.g., G418 for mammalian expression plasmid carrying the neomycin resistance gene). The day before transfection, seed PC12 cells at high density (at least 90-95% confluency) on collagen-coated tissue culture plates and maintain in complete growth medium.
- For PC12 cells plated on a 100-mm dish, add 8 µg DNA to 500µl OptiMEM-I in one tube (tube 1) and add 20µl Lipofectamine2000 to 500µl OptiMEM-I in another (tube 2). The contents of tube 2 should be incubated at room temperature for at least 5min before dropwise addition to tube 1. After which, incubat the mixture further at room temperature for 20min.
- During this time, remove complete growth medium from PC12 cells and replace with 4ml of OptiMEM-I.
- Add the Lipofectamine2000/DNA mixture to the PC12 cells and return the culture to the incubator. After 4-6 h, aspirate the Lipofectamine2000/DNA mixture (in OptiMEM-I) off completely and replace with complete growth medium. Expression of the transgene can be monitored 24-48 h afterward.
- Note that the amount of DNA and Lipofectamine2000, as well as the volume of OptiMEM-I, can be adjusted proportionally to accommodate PC12 cells that are plated on smaller/larger size tissue culture plates. The aforementioned procedure can also been used to transfect PC12 cells that were already differentiated by NGF treatment. However, the transfection efficiency is significantly lower (1-2%) due to the need to culture neuronally differentiated PC12 cells at lower density.
By adhering to the aforementioned protocols, our laboratory has been able to maintain (since 1977) PC12 cell stocks that are consistently responsive to NGF and that present a stable phenotype. However, a survey of the literature concerning the use of PC12 cells occasionally reveals conflicting or inconsistent results between laboratories. One possible cause for this may be the generation of variant "PC12 cell" lines. Like other continuous cell lines, PC12 cells are subjected to spontaneous mutations, and clonal PC12 cell variants have been identified from past studies. The introduction of nonstandard culture methods (e.g., changing sera, medium, substrate) can favor the selection of such variants over the wild-type population. Although the use of "variant" PC12 cell lines does not necessarily undermine the validity of data generated with them, it can give rise to uncertainty or confusion when one attempts to integrate/reproduce the finding from various reports. It is therefore our suggestion that a uniform standard of culturing PC12 cells be adopted for studies with this cell line.
Another cautionary note on the use of the PC12 cell line is that although it is a convenient model system for studying neuronal development and function, it is not a full substitute for "bona fide" nerve cells. Therefore, whenever feasible, experimental results obtained with PC12 cells should be verified or compared with representative neurons.
- Poor survival or growth of stock cultures has three probable causes: (i) the initial plating density is too low, (ii) the horse serum is not properly heat inactivated or is of insufficient quality (the latter is the usual cause for failure to thrive), and (iii) the culture medium is outdated (the glutamine has degraded).
- The most probable cause of poor cell adherence is an insufficient level of collagen as the substrate.
- A poor NGF response is indicated by the continuous proliferation of PC12 cells in NGF-containing medium and by the lack of neurite-bearing cells even after long-term NGF treatment. A possible cause is that the initial plating density is too high. Another is that the NGF may be inactive. Alternatively, the collagen concentration on the dishes is too high or too low or the collagen has deteriorated. Finally, the cultures may contain a high proportion of nonresponsive variants (in this case, start with a new cell stock of lower passage number).
- Spontaneously arising PC12 cell variants are indicated by the presence of flat (phase-dark), rapidly dividing, non-NGF responsive cells. Alternatively, contaminating variants may appear spiky in morphology even in the absence of NGE The best solution to this is to replace the entire stock with PC12 cells from an earlier passage and to adopt culture conditions that do not favor selection of variants.
- More than 50% of PC12 cells in serum-free medium without NGF should die within 24h after plating. However, PC12 cells at high density are capable of conditioning the culture medium, retarding death. Therefore, if cultures exhibit a delay in serumfree cell death, the experiment should be repeated with a lower density of cells. Alternatively, a delay of cell death could be due to an insufficient washout of serum or, for neuronally differentiated cultures, of NGE In this case, a more stringent washing procedure should be instituted.
- Generally, it is prudent to discard contaminated cultures and replace with fresh PC12 cell stock. However, if it is necessary to rescue a nonreplaceable culture (such as cell line established from transfection experiments), the following treatments may be effective in removing common sources of contamination. (i) Yeast: treat the culture with 1% fungizone (final concentration) in complete medium. (ii) Mold: remove the contaminant by aspiration. Alternatively, use a Pasteur pipette to remove some of the PC12 cells from a small unaffected area of the dish and replate the cells onto a new dish. Treat the cells with 1% fungizone in complete medium. (iii) Bacteria: a combination of antibiotics and bacterial static agents [see Sambrook et al. (1989) for appropriate doses] may be added to the culture. PC12 cells can tolerate ampicillin, kanamycin, spectinomycin, tetracycline, and chloramphenicol.
References
Greene, L. A., Aletta, J. M., Rukenstein, A., and Green, S. H. (1987). PC12 pheochromocytoma cells: Culture, nerve growth factor treatment, and experimental exploitation. Methods Enzymol. 147B, 207-216.
Greene, L. A., Sobeih, M. M., and Teng, K. K. (1991). Methodologies for the culture and experimental use of the PC12 rat pheochromocytoma cell line. In "Culturing Nerve Cells" (G. Banker and K. Goslin, eds.), pp. 207-226. MIT Press, Cambridge, MA.
Greene, L. A., and Tischler, A. S. (1982). PC12 pheochromocytoma cultures in neurobiological research. Adv. Celt. Neurobiol. 3, 373-414.
Kleitman, N., Wood, P. M., and Bunge, R. P. (1991). Tissue culture methods for the study of myelination. In "Culturing Nerve Cells" (G. Banker and K. Goslin, eds.), pp 337-377. MIT Press, Cambridge, MA.
Levi, A., and Alemái, S. (1991). The mechanism of action of nerve growth factor. Annu. Rev. Pharmacol. Toxicol. 31, 205-228.
Mobley, W. C., Schenker, A., and Shooter, E. M. (1976). Characterization and isolation of proteolytically modified nerve growth factor. Biochemistry 15, 5543-5552.
Sambrook, J., Fritsch, E. E, and Maniatis, T. (1989). "Molecular Cloning: A Laboratory Manual," 2nd Ed. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY.
Soto, A. M., and Sonnenschein, C. (1985). The role of estrogen on the proliferation of human breast tumor cells (MCF-7). J. Steroid Biochem. 23, 87-94.
Support our developers




