Embryonic Explants from Xenopus laevis as an Assay System to Study Differentiation of Multipotent Precursor Cells
I. INTRODUCTION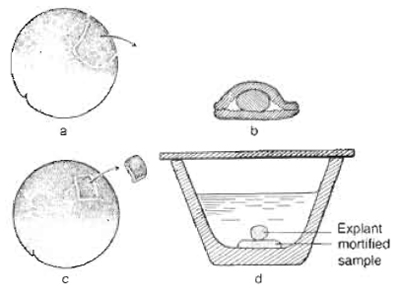 |
| FIGURE 1 Schematic representation of the "Umhfillungsversuch" (coating test) and "Auflagerungsversuch" (bedding test) by Johannes Holtfreter, 1933. (a) Two pieces of ectoderm were isolated using a self-made cutting tool (eyebrow). (b) The mortified implant was placed between ectodermal tissues. (c) A small piece of animal ectoderm of a gastrula stage embryo was excised (c) and placed on the mortified sample (d). Adapted from Spemann, 1936. |
Holtfreter showed that the mortified dorsal lip of a gastrula stage amphibian embryo led to the induction of neural tissue (neurale Blasen) in conjugated ectodermal explants (Fig. lb). In 1971, Sudarwati and Nieuwkoop demonstrated that vegetal tissue is capable of inducing mesoderm in combined animal cap explants using the South African clawed frog Xenopus laevis as a source for animal cap explants (mesoderm induction assay). More recently, the animal cap system has been utilized to tackle a broad variety of developmental problems, including the specification of all germ layers, organ formation, and expression screenings for secreted proteins (Sudarwati and Nieuwkoop, 1971; Gurdon et al., 1985; Slack, 1990; Smith et al., 1990; Green et al., 1992; Moriya et al., 2000).
Adult X. laevis frogs are from NASCO (Wisconsin). The reagents used are agarose (Cat. No. 15510-027, GibcoBRL); albumin bovine (BSA, Cat. No. A-8806, Sigma); chorionic gonadotropin (HCG, Cat. No. CG- 10, Sigma); L-15 (Leibovitz, GibcoBRL); L-cysteine hydrochloride monohydrate (Cat. No. 30129, Fluka); HEPES (Cat. No. H-3375, Sigma); and penicillin/ streptomycin solution (with 10,000 units penicillin and 10mg streptomycin/ml, Cat. No. P-0781, Sigma). All other chemicals used are from Merck.
The gastromaster and replacement microsurgery tips are from XENOTEK Engineering (Belleville, IL). The microinjector for RNA injection is from Eppendorf (Microinjector 5242, Eppendorf).
III. BASIC PROCEDURES
The basic principles of the procedure have been described previously (Grunz and Tacke, 1989; Hollemann et al., 1998). In addition to an update of the basic protocol, this article describes several readout systems that can be applied.
Solutions
Modified Barth's solution (MBS): 88mM NaCI, 2.4 mM NaHCO3, 1.0 mM KCI, 10 mM HEPES, 0.82mM MgSO4, 0.41mM CaCl2, and 0.33mM Ca(NO3)2, pH 7.4
Ca2+M//Mg2+-free MBS: 88mM NaCl, 2.4mM NaHCO3, 1.0mM KCl, and 10mM HEPES, pH 7.4
L-15/BSA solution: 65% L-15 and 0.1% BSA, pH 7.4
Histone H4 primer: H4-F: 5'-CGGGATAACATTC AGGGTATCACT-3' and H4-R: 5'-ATCCATGGCG GTAACTGTCTTCCT-3'
RT mix (one reaction; 5µl): 1 µl 25 mM MgCl2 solution, 0.5 µl 10× PCR buffer II, 2 µl 2.5 mM dNTPs, 0.25 µl RNase inhibitor (20U/µl), 0.25µl MuLV reverse transcriptase (RT) (50U/µl), 0.25µl 50µM random hexamers, and 0.75µl (35 ng) RNA sample
PCR(RT) mix (one reaction; 20ml): 0.5 ml 25 mM MgCl2 solution, 2ml 10× PCR buffer II, 16.625 ml RNasefree H2O, 0.375ml 10mM forward primer, 0.375 ml 10mM reverse primer, and 0.125 ml Ampli Taq DNA polymerase (5 U/ml)
At the late blastula stage (stage 8.5-9), Xenopus embryos show a relatively defined segregation of presumptive ectoderm (animal pole), mesoderm (equatorial area), and endoderm (vegetal pole); the blastocoel has attained its full size, and the inner surface of the blastocoel becomes smooth. The roof of the blastocoel, the animal cap (very top of the animal pole), consists of a single outer layer and two inner layers of cells. The pigmentation pattern and the cell size differences make it possible to roughly distinguish the presumptive ectoderm, mesoderm, and endoderm from outside of the embryo. However, the easiest way to identify the animal pole (animal cap) is to look at the embryos directly. What you see from the top is the animal pole, as the embryos are naturally floating inside the vitelline membrane with the animal pole up and the vegetal pole down due to gravity.
Preparation of Xenopus Embryos
Eggs from adult Xenopus are obtained from females 6-8h after injection with human chorionic gonadotropin (500-1000 U/frog) and fertilized with minced testis in 0.1× MBS. Forty minutes after fertilization, dejelly the embryos in 2% cysteine (pH 8.0) and wash extensively in 0.1× MBS.
As there is no marker to clearly delineate the boundary of presumptive ectoderm and mesoderm in a living embryo, it is difficult to isolate animal caps without presumptive mesodermal cell contamination. Many tools, including forceps, hair loops, fine glass needles, and tungsten needles, have been developed for cutting animal caps. The most advanced tool is the gastromaster. No matter which tool is used, the first step is to manually remove the vitelline membrane of stage 8.5-9 embryos (the best stage for capping) with a pair of watchmaker's forceps in a petri dish coated with 1% agarose. Although the manufacturer of the gastromaster demonstrates that it is possible to directly isolate animal caps without removing the vitelline membrane (see video clips at http://www.gastromaster.com/), we strongly recommend removing the membrane manually. The forceps do not have to be very sharp, but the two tips should be well matching. Digging a small pit into the surface of the coated agarose is helpful for fixing the embryo during manipulation. It is recommended to hold and remove the vitelline membrane with forceps from the equatorial area or from the vegetal pole, thus avoiding damage to the animal pole cells. Once the embryos are released from the vitelline membrane, they can no longer rotate freely according to gravity. Therefore, in order to isolate a clean animal cap from the right animal pole region, it is extremely important to properly orientate the nude embryo animal pole up and vegetal pole down (Fig. 2a). Dissect the animal cap (very top region of the animal pole, illustrated by the dashed circle in Fig. 2a) in 0.5× MBS using a gastromaster equipped with a yellow microsurgery tip of 400-500 mm in width (Figs. 2b-2e). This step is quite easy compared to the removal of vitelline membrane. We encourage visiting the gastromaster manufacturer's website for video clips that nicely demonstrate the dissecting process. Culture animal caps in 0.5× MBS containing penicillin (100U/ml) and streptomycin (100µg/ml) and harvest until control siblings have reached desired developmental stages for further analyses.
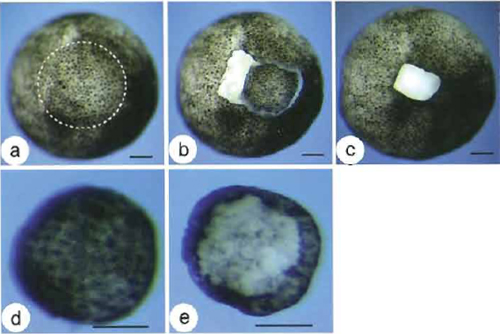 |
| FIGURE 2 Cutting an animal cap with the gastromaster. (a) Animal pole view of a demembranated stage 9 Xenopus embryo. The dashed circle demarcates the very top region of the animal pole (the animal cap or presumptive ectoderm). (b) An animal cap freshly dissected with the gastromaster, still sitting on the mother embryo. (c) Wound after the isolation of the animal cap. (d) Outer layer view of an isolated animal cap. (e) Inner layer view of an isolated animal cap. Bars: 100µm. |
B. Manipulation of Animal Cap Cells
RNA Injection Techniques
One approach to investigate the activity of individual proteins is to introduce the corresponding mRNAs into the animal pole of early cleavage stage embryos followed by the isolation of animal caps for further analysis. Depending on the gene of interest, 5-2000 pg, e.g., FGF or XCYP26 (Isaacs et al., 1992; Hollemann et al., 1998), of in vitro-synthesized capped mRNA (Mega Message Machine, Ambion, USA) in 10nl RNase-free water can be microinjected into the animal pole of both blastomeres of two-cell stage embryos, which are transferred into l× MBS and kept at 13°C to slow down development during injection. Two hours after injection, culture embryos again in 0.1× MBS.
Application of Growth Factors
An alternative approach to monitoring the inductive activities of growth factors is to directly apply the active form of a given growth factor protein to the isolated animal caps with or without dissociation. The advantage of dissociation is that each cell is exposed to a uniform factor concentration.
Effects on gene transcription can be analyzed by RTPCR and whole mount in situ hybridization.
RT-PCR Analysis
The animal cap system can be used to elucidate which factors and signal transduction pathways regulate genes of interest or, vice-versa, which genes are downstream targets. Effects on gene transcription in explants can be analysed either by RNase protection or RT-PCR. We use a nonradioactive RT-PCR assay. It is normally sufficient to use this simple, semiquantitative approach to analyse strong effects on gene expression. A variety of other RT-PCR methods have also been described for Xenopus: quantitative, radioactive RTPCR (Rupp and Weintraub, 1991) and quantitative, competitive RT-PCR (Onate et al., 1992). The most sensitive and most reproducible technique based on fluorescence kinetics is quantitative real-time RT-PCR (Xanthos et al., 2002), but it requires a special thermocycler.
RNA Isolation from Animal Cap Explants
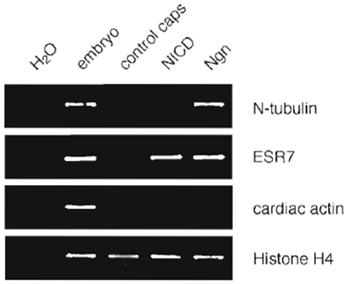 |
| FIGURE 3 Induction of neuronal gene expression in animal caps as analyzed by RT-PCR. One hundred picograms X-Ngnr-1 mRNA (Ngn) or 300 pg Notch ICD mRNA (NICD) was injected in both cells of two-cell stage embryos, animal caps were dissected at early stage 9, cultured to stage 15, and then analyzed by RT-PCR. RNA isolated from uninjected total embryos of stage 15 was used as a positive control. X-Ngnr-1 induces the neuronal differentiation marker N-tubulin, as well as the Notch target gene ESR7 in animal caps. Histone H4-specific primers were used to control RNA input and quality. As shown by the absence of transcripts for cardiac actin, the isolated explants were not contaminated by mesodermal tissue (S61ter, et al., unpublished results). |
We use the RNeasy minikit (Qiagen) to isolate RNA from animal caps and whole embryos for RT-PCR analysis following the RNeasy miniprotocol for the isolation of total RNA from animal tissues (see manufacturer's instructions with modifications detailed later). Digest genomic DNA using DNase I (Qiagen) for on column treatment according to the manufacturer's protocol.
Analysis of Genomic DNA Contamination
The exon-intron structure of most Xenopus genes is unknown. Therefore, primers used for the analysis that can target the same exon and genomic DNA contamination of isolated RNA would thus result in falsepositive signals. In order to control for genomic DNA contamination, a regular PCR reaction for histone H4 genomic DNA is performed. Add 1 µl RNA (50ng/µl) to 25µl PCR mix containing the histone H4-specific primer. Use genomic DNA (5 ng) as a positive control and H2O as a negative control, with the following cycling parameters: 94°C 2 min (94°C 45 s, 58°C 45 s, 72°C 30s) × 35, 72°C 5 min. Analyze one-half of the PCR on a 2% TBE agarose gel.
RT-PCR
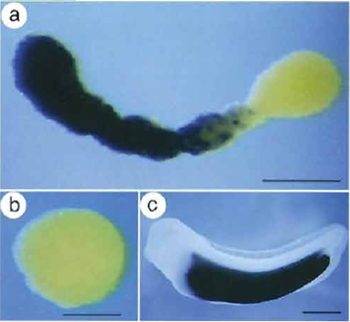 |
| FIGURE 4 Induction of endodermal gene expression in animal cap explants as analysed by whole mount in situ hybridisation. Expression analysis of the activation of an endoderm-specific gene in VegT/β-catenin-injected animal cap explants. Cy118 encodes a novel putative peptidase that demarcates the developing embryonic intestine. VegT (500pg/embryo) and β-catenin (200pg/embryo) mRNAs were injected into the animal pole of both blastomeres of two-cell stage embryos. Animal caps were isolated from stage 9 control uninjected and VegT/β-catenin-injected embryos and collected when control siblings had reached stage 34. (a) Cy118 expression is activated in a subset of cells of an elongated animal cap injected with VegT/β-catenin. (b) Cy118 is not expressed in the control cap. (b) Cy118 is expressed exclusively in the presumptive intestine of stage 34 embryos (Chen et al., 2003). |
Whole Mount in Situ Hybridization
Gene expression on the RNA level in animal cap explants can also be analyzed by whole mount in situ hybridization, originally adapted for Xenopus embryos by Harland (1991). The advantage of this method in comparison to RT-PCR is that individual explants can be controlled for homogeneity of gene expression on the level of individual cells. The disadvantage is that substantial numbers of explants are required if several genes are to be analyzed in parallel. We use the basic protocol for whole mount in situ hybridization described previously (Hollemann et al., 1999) with the following minor modifications. In case of several samples, a transfer of fixed animal caps from glass vials to 24-well tissue culture plates before rehydration facilitates handling. Proteinase K treatment should be shortened to 5-8 min at room temperature.
RT-PCR Analysis of Animal Cap Explants
Many different genes are involved in the activation of the neuronal differentiation program, which drives naive ectodermal cells to become postmitotic neurons. In animal cap explants, X-ngnr-1 can also promote neurogenesis, as monitored by the activation of the neural determination marker N-tubulin. The density of cells expressing endogenous X-ngnr-1 is controlled by lateral inhibition mediated through Notch signalling (Ma et al., 1996). In response to misexpression of the intracellular domain of Notch (ICD) that mimics active Notch signalling, the expression of target genes, such as Esr7, is induced (Fig. 3).
 |
| FIGURE 5 Elongation of animal caps mimics gastrulation movements. Animal cap explants of Xenopus laevis were isolated at stage 8 and treated with activin for 2h. Treated caps elongate until stage 18 (left). Arrows indicate examples for elongation of the caps. In this example, all animal caps shown have indications of elongation. Untreated control caps remain spherical (right). |
In addition to neural and mesodermal tissue, animal cap explants can also be converted into endoderm. For instance, VegT can synergize with β-catenin to induce a number of liver- and intestine-specific genes in animal cap explants. Figure 4 shows an example of whole mount in situ hybridization analysis for the induction of an endoderm marker gene in an animal cap upon VegT/β-catenin injection.
The Animal Cap to Study Cell Migration
Upon treatment with activin or FGF, the animal cap is not only induced to become mesoderm (see above), but as a consequence of mesoderm induction, the animal cap also changes its form (Fig. 5). Whereas untreated animal caps round up and look like spherical balls, induced animal caps elongate as a consequence of cell movements within the animal cap (Kuhl et al., 2001). It is widely accepted that this kind of cell movement within the cap mimics mesodermal cell movements normally occurring during gastrulation of the Xenopus embryo. During gastrulation, cells from a more ventral position migrate towards the dorsal midline. The forces generated by this process in vitro not only push cells during gastrulation over the dorsal lip, but also lead to an elongation of the embryo along its anterior-posterior axis (for a review on cell movements during gastrulation, see Keller, 1986). With respect to gastrulation movements, the animal cap thus serves as an excellent system to study the molecular basis of cell migration. The elongation behaviour of injected or growth factor-treated caps can be studied and conclusions can be drawn on the function of a gene of interest with respect to cell migration. As an example, we show the effect of activin on animal caps (Fig. 5).
Recapitulation of Organ Formation in Animal Caps
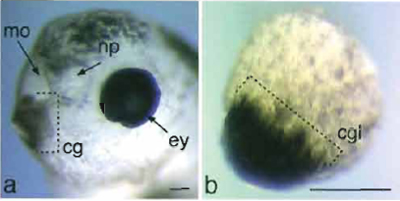 |
| FIGURE 6 Cement gland formation in animal cap explants. (a) Anterior to the left, sloped lateral view of a head of a noninjected Xenopus larvae at NF stage 40. The cement gland is positioned ventrally immediately adjacent to the mouth opening. (b) Example of an animal cap explant that had been injected with Xpitxl into one cell at the two-cell stage and cultured equivalent to stage 40. Bar: 50µm. cg, cement gland; cgl, cement gland like; ey; eye; mo, mouth opening; np, nasal pit. |
b. Notochord from Dispersed Animal Cap Explant Cells. In response to different concentrations of activin, dispersed animal cap cells have been shown to exhibit sharp thresholds in respect to target gene activation. Activin A is added to the cells at a final concentration of 4 ng/ml for notochord differentiation and 8 ng/ml for endoderm differentiation and incubated at room temperature for 1 h. In most cases, more than 90% of the reaggregated tissue mass from each batch of preparation differentiated into notochords. Some neural cells were formed concomitantly.
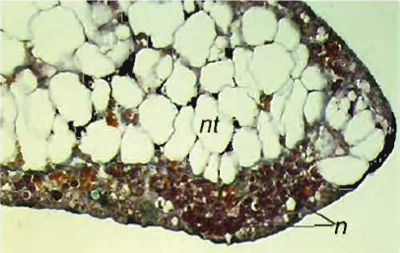 |
| FIGURE 7 Histological staining of notochord tissues generated from animal cap cells treated with activin. In order to judge the inductive activities, one piece of the tissues from each batch of experiments was cultured further and finally fixed in Bouin's solution when control embryos reached stage 42. Differentiation of the tissues is assayed by histological analysis, nt, notochord cells; n, neural cells. |
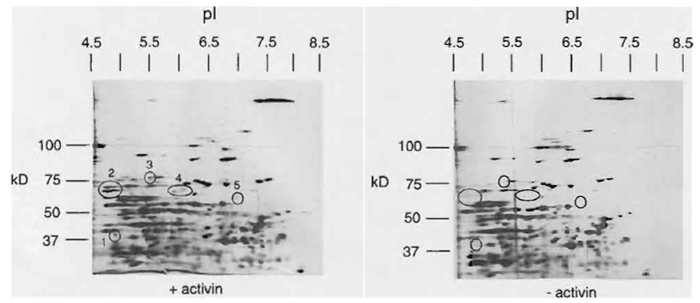 |
| FIGURE 8 Two-dimensional gel analysis of proteins expressed in untreated and activin-treated animal caps. Embryos were injected at the two-cell stage with activin mRNA. Animal caps were isolated at stage 8 and cultured until stage 19. Equivalents of four animal caps were used for 2D protein gels. Circles indicate spots of different intensity upon treatment. Whereas spots I to 3 most likely represent proteins that are upregulated through activin treatment, spots 4 and 5 indicate two spots that are weakly affected and might also represent artefacts. Two-dimensional gels with a broad IP range were used. For a more detailed analysis of inductive processes, small range IP gels are required to increase the resolution. See detailed literature on 2D gel analysis for methods and detailed interpretation of the 2D gel. (Shevchenko et al., 1996). |
References
Chen, Y., Jurgens, K., Hollemann, T., Claussen, M., Ramadori, G., and Pieler, T. (2003). Cell-autonomous and signal-dependent expression of liver and intestine marker genes in pluripotent precursor cells from Xenopus embryos. Mech. Dev. 120, 277-288.
Green, J. B., New, H. V., and Smith, J. C. (1992). Responses of embryonic Xenopus cells to activin and FGF are separated by multiple dose thresholds and correspond to distinct axes of the mesoderm. Cell 71, 731-739.
Gurdon, J. B., Fairman, S., Mohun, T. J., and Brennan, S. (1985). Activation of muscle-specific actin genes in Xenopus development by an induction between animal and vegetal cells of a blastula. Cell 41, 913-922.
Harland, R. M. (1991). In situ hybridization: An improved wholemount method for Xenopus embryos. Methods Cell Biol. 36, 685-695.
Hollemann, T., Chen, Y., Grunz, H., and Pieler, T. (1998). Regionalized metabolic activity establishes boundaries of retinoic acid signalling. EMBO J. 17, 7361-7372.
Hollemann, T., Panitz, E, and Pieler, T. (1999). In situ hybridization techniques with Xenopus embryos. In "A Comparative Methods Approach to the Study of Oocytes and Embryos" (J. D. Richter, ed.), pp. 279-290. Oxford Univ. Press, New York.
Hollemann, T., and Pieler, T. (1999). Xpitx-l: A homeobox gene expressed during pituitary and cement gland formation of Xenopus embryos. Mech. Dev. 88, 249-252.
Holtfreter, J. (1933). Nachweis der Induktionsf~ihigkeit abget6teter Keimteile. Isolations- und Transplantationsversuche. Roux" Arch. 128, 585-633.
Isaacs, H. V., Tannahill, D., and Slack, J. M. (1992). Expression of a novel FGF in the Xenopus embryo: A new candidate inducing factor for mesoderm formation and anteroposterior specification. Development 114, 711-720.
Keller, R. E. (1986). The cellular basis of amphibian gastrulation. Dev. Biol. 2, 241-327.
Kuhl, M., Geis, K., Sheldahl, L. C., Pukrop, T., Moon, R. T., and Wedlich, D. (2001). Antagonistic regulation of convergent extension movements in Xenopus by Wnt/beta-catenin and Wnt/Ca2+ signaling. Mech. Dev. 106, 61-76.
Moriya, N., Komazaki, S., Takahashi, S., Yokota, C., and Asashima, M. (2000). In vitro pancreas formation from Xenopus ectoderm treated with activin and retinoic acid. Dev. Growth Differ. 42, 593-602.
Onate, A., Herrera, L., Antonelli, M., Birnbaumer, L., and Olate, J. (1992). Xenopus laevis oocyte G alpha subunits mRNAs: Detection and quantitation during oogenesis and early embryogenesis by competitive reverse PCR. FEBS Lett. 313, 213-219.
Rupp, R. A., and Weintraub, H. (1991). Ubiquitous MyoD transcription at the midblastula transition precedes induction-dependent MyoD expression in presumptive mesoderm of X. laevis. Cell 65, 927-937.
Shevchenko, A., Jensen, O. N., Podtelejnikov, A. V., Sagliocco, E, Wilm, M., Vorm, O., Mortensen, P., Boucherie, H., and Mann, M. (1996). Linking genome and proteome by mass spectrometry: Large-scale identification of yeast proteins from two dimensional gels. Proc. Natl. Acad. Sci. USA 93, 14440-14445.
Slack, J. M. (1990). Growth factors as inducing agents in early Xenopus development. J. Cell Sci. Suppl. 13, 119-130.
Smith, J. C., Price, B. M., Van Nimmen, K., and Huylebroeck, D. (1990). Identification of a potent Xenopus mesoderm-inducing factor as a homologue of activin A. Nature 345, 729-731.
Sudarwati, S., and Nieuwkoop, P. D. (1971). Mesoderm formation in the anuran Xenopus laevis (Daudin). Roux' Arch. 166, 189-204.
Xanthos, J. B., Kofron, M., Tao, Q., Schaible, K., Wylie, C., and Heasman, J. (2002). The roles of three signaling pathways in the formation and function of the Spemann organizer. Development 129, 4027-4043.
Support our developers




