TERA2 and Its NTERA2 Subline: Pluripotent Human Embryonal Carcinoma Cells
TERA2 is one of the oldest extant cell lines established from a human teratocarcinoma. It was derived from a lung metastasis of a testicular germ cell tumour and reported by Fogh and Tremp in 1975. However, its pluripotent embryonal carcinoma (EC) properties were not immediately obvious, in part because maintenance of its undifferentiated EC phenotype requires that cultures of these cells are passaged by scraping in order to retain small clumps of cells instead of using trypsinisation, which results in single cell suspensions. In an early study of human teratocarcinoma-derived cell lines we ourselves dismissed TERA2 as a potential EC cell line (Andrews et al., 1980).
Initially, we failed to derive xenograft tumours from TERA2 (Andrews et al., 1980). Nevertheless, after a number of attempts we did succeed in obtaining a single xenograft tumour after injecting a nude, athymic (nu/nu) mouse with TERA2 cells. Fortunately, we explanted some of this tumour back into culture, as well as fixing part for histology. To our surprise, histological examination then revealed that the tumour contained a variety of differentiated elements, as well as embryonal carcinoma components; indeed it was a teratocarcinoma. Meanwhile, it was evident that the cells in culture, now named NTERA2 to designate their passage through a nude mouse, did not resemble morphologically the cells that predominated in the culture of TERA2 that had been used to inoculate the host mouse (Andrews et al., 1980). Indeed, the NTERA2 cells closely resembled other cells that we believed to represent human EC cells (Andrews et al., 1982, 1984b).
Several single cell clones of NTERA2 were initially studied, in particular NTERA2 clone B9, clone D3, and clone D1. However, there was no obvious difference among these and most subsequent studies have utilised the clone NTERA2 cl. D1, which has also been abbreviated NT2/D1 or sometimes simply NT2. The different clones do exhibit slightly different karyotypes, and it is certain that a small amount of genetic drift occurs upon prolonged culture. Nevertheless, the modal chromosome numbers of the NTERA2 clones, and of TERA2 itself, are very similar, about 61 chromosomes, including a variety of rearrangements. Many of these rearrangements are common to all clones, but new rearrangements continue to appear and characterise the individual clones (Andrews et al., 1984b, 1985).
The features of undifferentiated human EC cells are best exemplified by another cell line, derived from a testicular germ cell tumour, 2102Ep (Andrews et al., 1982). 2102Ep cells, and indeed a number of other "nullipotent" human EC cell lines (Andrews et al., 1980; Andrews and Damjanov, 1994), tend to grow in tightly packed colonies of small cells with little cytoplasm and few prominent nucleoli within a nucleus that comprises most of the cell. They exhibit doubling times of about 20-24 h, and eventually form monolayers from which domes may form and floating vesicles bud off. The cells of these vesicles do not seem to be significantly different from cells that remain attached to the substrate. However, high-density cultures are required to retain these featuresmtypically we seed cultures at densities of at least 5 × 106 cells per 75-cm2 flask. At lower densities, the cells begin to flatten out and evidently some differentiate into trophectoderms (Andrews et al., 1982; Andrews, 1982; Damjanov and Andrews, 1983).
NTERA2 EC cells behave similarly, although there are differences. Maintenance at high cell densities is essential but, in this case, for passaging, cultures should be dispersed by scraping instead of using trypsin and EDTAmcell:cell contact seems to be required for the long-term retention of an undifferentiated phenotype. Like 2102Ep, NTERA2 cells at low density also flatten out and appear to differentiate, e.g., inducing expression of fibronectin, but they do not seem to produce trophectoderm (Andrews, 1982; Andrews et al., 1984b). Extraembryonic endoderm, evidenced by the production of laminin, may be formed (Andrews et al., 1983).
In these respects, human EC cells differ from their murine counterparts, which do not express SSEA3 or SSEA4, or murine Thy-1 (Shevinsky et al., 1982; Kannagi et al., 1983; Martin and Evans, 1974), but do express SSEA1 (Solter and Knowles, 1978). Murine EC cells do express high levels of alkaline phosphatase (Bernstine et al., 1973), but that is not recognised by the antibodies TRA-2-49 and TRA-2-54. The antibodies TRA-1-60, TRA-1-81, and GCTM2 also do not appear to recognise epitopes expressed by mouse cells. A further difference between mouse and human EC cells is the expression of class 1 major histocompatibility complex (MHC) antigens, of which HLA is commonly expressed by human EC cells (Andrews et al., 1981), whereas H-2 is not expressed by mouse EC cells. In fact, undifferentiated NTERA2 cells only express low and variable levels of HLA-A,B,C, in contrast to other human EC cells (Andrews et al., 1984b), but these levels are increased markedly by exposure to interferon- 1 (Andrews et al., 1987).
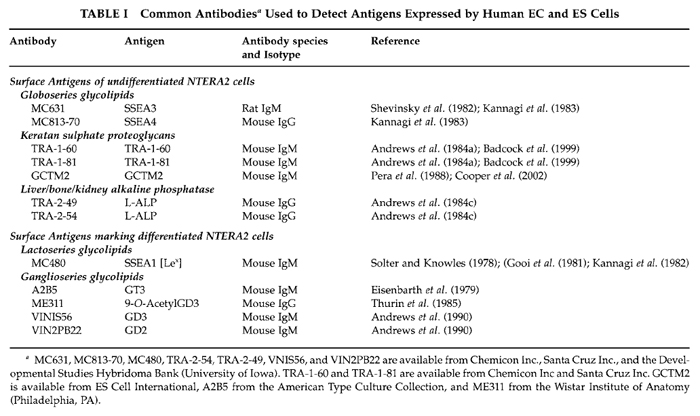 |
NTERA2 EC cells differ from many other human EC cells by their susceptibility to differentiation induced by retinoic acid (Andrews, 1984) and to other inducing agents such as hexamethylene bisacetamide (HMBA) (Andrews et al., 1986, 1990) and the bone morphogenetic proteins (Andrews et al., 1994). These agents induce differentiation in distinct directions, although the best studied is retinoic acid-induced differentiation, which results in the formation of neurons as well as other cell types (Andrews, 1984). By contrast, many other human EC cells do not respond to retinoic acid (Matthaei et al., 1983).
Within 24-48h after exposure to 10-5M all-trans retinoic acid, NTERA2 cells commit to differentiate. During the following 2 weeks, expression of the key surface markers SSEA3, SSEA4, TRA-1-60, and TRA-1- 81 is eliminated in most cells, while they lose the characteristic EC morphology, and begin expressing a range of new markers. Prominent among these induced markers are surface antigens SSEA1, A2B5, and ME311 (Table I), which appear to segregate to discrete subsets of cells (Fenderson et al., 1987). At the same time, expression of members of HOX gene clusters is induced (Mavilio et al., 1988). This induction of HOX gene expression is dose dependent in a way that relates to the position of the genes in the HOX cluster (Simeone et al., 1990; Bottero et al., 1991). Thus, genes located at the 5' end of the clusters tend to require higher concentrations of retinoic acid for maximal induction than genes located at the 3' end of the clusters. This pattern appeared to relate to the expression pattern of the HOX genes along the anterior-posterior axis of the developing embryo and the postulated role for retinoic acid in establishing that axis. However, differentiation induced by HMBA, which causes a similar elimination of the EC surface marker antigens, was not accompanied by a comparable induction of the surface antigens, or the HOX genes, induced by retinoic acid (Andrews et al., 1990).
Among cells arising from the differentiation of NTERA2, the neurons that appear after retinoic acid induction are the most well defined and studied. These appear only rarely after induction with HMBA or BMPs (Andrews et al., 1986, 1990, 1994). A variety of other cell types do appear in cultures induced with each of these agents. These are poorly defined and are not identified readily with specific cell types, although they may include mesenchymal cell types, including smooth muscle, fibroblasts, and chondrocytes (Duran et al., 1992).
Medium
All cells are cultured in Dulbecco's modified Eagle's medium (DMEM) (high glucose formulation), supplemented with 10% fetal calf serum (FCS). DMEM may be purchased from any reputable supplier of tissue culture reagents. Samples of FCS should be obtained from a number of suppliers and tested to identify a batch that supports optimal growth of NTERA2 cells and maintenance of an undifferentiated state, assayed by the expression of appropriate markers (see later). A batch of FCS that provides for good maintenance of undifferentiated NTERS2 cells is not necessarily the best for supporting differentiation. FCS should be batch tested separately for both purposes.
Dulbecco's Phosphate-Buffered Saline (PBS)
For the following procedures, PBS without Ca2+ and Mg2+ is used and may be purchased from any reputable supplier of tissue culture reagents.
all-trans Retinoic acid can be purchased from Sigma- Aldrich or from Eastman-Kodak. It should be stored in the dark at -70°C, preferably under nitrogen. A stock solution of 10-2M retinoic acid (3mg/ml) in dimethyl sulphoxide (DMSO) should be prepared and also stored at-70°C. If prepared carefully under asceptic conditions, this stock solution may be assumed to be sterile.
Hexamethylene Bisacetamide (HMBA)
A 0.3 M stock solution of HMBA should be prepared in PBS and may be stored at 4°C. It may be sterilised by passage through a 0.2-µm filter.
Glass Beads
Purchase 3mm flint glass beads from a reputable laboratory supplier. These should be washed in 10N HCl and rinsed thoroughly with water. After drying, about 20 should be placed in a Wasserman tube and sterilised by autoclaving.
Trypsin:EDTA
A solution of 0.25% trypsin in 1 mM EDTA and PBS (Ca2+/Mg2+ free) may be purchased from any reputable tissue culture supplier.
Mitotic Inhibitors
- Cytosine arabinoside: Prepare a fresh 1 mM stock solution every 2 weeks and store at 4°C.
- Fluorodeoxyuridine: Store a 1 mM stock solution in water at-20°C.
- Uridine: Store a 1 mM stock solution in water at -20°C.
Matrigel is a proprietary product of Invitrogen and consists of a mixture of extracellular matrix components produced by an established tumour cell line. It gels spontaneously at 37°C, but remains as a liquid at 4°C. It should be stored at-20°C on receipt. To use, thaw a vial by standing on ice overnight. The thawed Matrigel may be distributed into convenient aliquots and refrozen once for further storage. To coat tissue culture surfaces, the thawed Matrigel should be diluted 1:30 in cold medium and enough to cover the surface should be pipetted into the desired tissue culture vessel (e.g., 1.5ml per 25-cm2 tissue culture flask), which should then be incubated at 37°C for 2 h. The Matrigel may then be aspirated and the flask is ready for plating cells.
V. PROCEDURES
A. Maintenance of NTERA2 Stock Cultures
NTERA2 cells should be maintained at high cell densities in DMEM plus 10% FCS at 37°C under a humidified atmosphere of 10% CO2 in air. The FCS should be batch tested to ensure that it is suitable for supporting optimal growth of the cells (population doubling times of approximately 20 h) in an undifferentiated state, as assessed by morphology and expression of surface markers (SSEA3, SSEA4, TRA-1-60, and TRA-1-81).
The cells should be harvested for subcultivation by scraping, which can be achieved most easily with the use of 3-mm-diameter glass beads, stored in Wasserman tubes. The contents of the tube may then be tipped directly into a flask of cells to affect the detachment of adherent cells as described.
- Choose a 75-cm2 flask of a well-grown, subconfluent culture of NTERA2 cells (if harvested as a single cell suspension, such a culture would yield about 20 × 106 cells).
- Aspirate some of the medium, leaving about 10ml behind (alternatively, remove all the medium and add 10 ml fresh medium).
- Tip about 20-30 sterile, acid-washed 3-mm glass beads into the flask and roll them over the surface of the flask. Rolling should be just vigorous enough that the adherent cells detach in clumps.
- Gently triturate the detached cell clumps to reduce their size to the order of 10-50 cells and transfer approximately one-third of these to a new flask, to which 20ml fresh medium should be added.
- The resulting culture should be maintained at 37°C under a humidified atmosphere of 10% CO2 in air; it should be subcultivated as just described after about 3-4 days.
B. Cryopreservation and Recovery of NTERA2 Cells
NTERA2 cells may be frozen in a freezing mixture consisting of 10% DMSO and 90% FCS.
Steps
- Harvest a well-grown, healthy culture of NTERA2 by scraping as described earlier.
- Centrifuge the cell suspension at 1000 rpm for 5 min.
- Remove the supernatant and resuspend the cells in freezing mixture: use about 0.6ml for cells harvested from a 75-cm2 flask.
- Distribute about 0.2ml of cell suspension in freezing mixture to screw-cap cryovials and transfer, in a cardboard box, to a -70°C freezer overnight.
- The next day, transfer the frozen vials of cells to a liquid nitrogen freezer.
- Thaw the cell suspension rapidly in a water bath at 37°C.
- Dilute the cell suspension in 5 ml medium and centrifuge at 1000 rpm for 5 min.
- Resuspend the cell pellet in fresh medium and transfer to a new 75-cm2 flask; culture at 37°C as before.
C. Differentiation of NTERA2
NTERA2 may be induced to differentiate by a variety of agents. However, the most widely used agents are all-trans retinoic acid and HMBA. The following protocol is used for the preparation of purified neural cells from retinoic acid-induced cultures of NTERA2 and is based upon the procedures of Andrews (1984) and Pleasure et al. (1992).
Steps
- Harvest cells from a stock culture using trypsin:EDTA and reseed at 1 × 106 cells per 75-cm2 tissue culture flask in 15 ml DMEM (high glucose formulation) with 10% FCS and 10 -5M all-trans retinoic acid diluted from the stock solution (10 -2 M) in DMSO. [Note: 10-5M retinoic acid is a relatively high concentration; lower concentrations may be used but certainly these have different effects, for example, with respect to induction of Hox genes (Simeone et al., 1990); also serum acts to buffer the concentration of free retinoic acid, and lower concentrations are necessary if used in conjunction with serum-free conditions.]
- Maintain the cultures at 37°C and refeed with fresh medium containing 10-5M retinoic acid every 7 days. [Note: Our standard protocol is to maintain the differentiating cells continuously in retinoic acid. However, it is possible to remove the retinoic acid after 2-3 days and achieve full differentiation of the cells with the formation of neurons, but there may be differences in the properties of the derivative cells.]
- After 3 weeks harvest the cells with trypsin:EDTA and reseed in a fresh flask at a split ratio of 1:2 in DMEM with 10% FCS but no retinoic acid. This is called "replate 1.'
- After 2-3 days add trypsin:EDTA (1 ml per 75- cm2 flask) and observe under the microscope. As the cells on the surface begin to detach (about I min), hit the side of the flask sharply with the flat of your hand to dislodge the loosely adhering, neural precursor cells. Dilute the detached cells in fresh medium. [Note: The object is to begin to purify the neural precursors from the other differentiated derivatives, which adhere to the substrate more strongly. Therefore, do not leave the trypsin:EDTA on the cells long enough to cause all the cells to detach, i.e., less than 3min.]
- Combine the harvested cells from several flasks and reseed at 6-8 × 106 cells per 25-cm2 flask in DMEM with 10% FCS and inhibitors (10-6M cytosine arabinoside, 10-5M fluorodeoxyuridine, 10-5M uridine). This is called "replate 2.' Feed these cultures with fresh medium including inhibitors every 3-4 days).
- After 1-2 weeks, treat the cultures with trypsin:EDTA (1 ml per 25-cm2 flask). Monitor the cultures under a microscope and preferentially remove the neurons that detach first (as in step 4). Dilute the suspended detached neurons in 10ml fresh medium and replate onto surfaces coated with Matrigel (see later) at a density of about 105 cells/cm2, as required for further experiments. The resulting neuronal cultures (called "replate 3")can be maintained for several weeks with refeeding every 4-7 days.
Monoclonal antibodies that are often used to assess surface antigens on undifferentiated NTERA2 cells and during their differentiation are shown in Table I. These antibodies are generally available as culture supernatants, purified antibodies, or ascites. They may be used in a variety of immunoassays, but indirect immunofluorescence, detected by flow cytometry, is especially useful. The latter can be readily adapted for sorting viable populations of cells for further functional or developmental studies (e.g., Ackerman et al., 1994; Pryzborski et al., 2000). Whichever preparation that is available should be pretitred on cells known to express the relevant antigen. As a negative control, we use the antibody from the original parent myeloma of most hybridomas, namely P3X63Ag8 (Kohler and Milstein, 1975). However, others may prefer a classmatched nonreactive antibody if one is available or else no first antibody at all.
Steps
- Prepare a single cell suspension by harvesting cells with trypsin:EDTA. After pelleting, resuspend the cells in HEPES-buffered medium or wash buffer (PBS plus 4% FCS and 0.1% sodium azide) at 2 × 106/ml.
- Choose antibodies and dilute as appropriate in wash buffer.
- Distribute the diluted antibodies at 50 µl per well of a round-bottom 96-well plate.
- Add 50 µl of cell suspension (i.e. 105 cells) to each 50 µl of antibody.
- Seal the plate by covering with a sticky plastic cover, ensuring that each well is sealed, and incubate at 4°C, with gentle shaking, for 30-60rain.
- Spin the plate at 280g for 3 min using microtitre plate carriers in a tissue culture centrifuge. Check that the cells are pelleted and remove the plastic seal using a sharp motion but holding the plate firmly to avoid disturbing the cell pellet. Dump the supernatant by inverting the plate with a rapid downward movement; blot the surface and turn the plate over. Provided that this is done in a single movement without hesitation, the cells remain as pellets at the bottom of the wells. If there are any concerns about pathogens contaminating a culture, supernatants can be removed by aspiration rather than dumping.
- Wash the cells by adding 100µl wash buffer to each well, seal, and agitate to resuspend the cells. Spin down as described earlier. After removing the supernatant, repeat with two further washes.
- After the third wash, remove the supernatant and add 50µl fluorescent-tagged antibody, previously titred and diluted in wash buffer to each well. FITCtagged goat antimouse IgM or antimouse IgG, as appropriate to the first antibody, may be used. Antimouse IgM, but not antimouse IgG, usually works satisfactorily with MC631 (a rat IgM). Affinity-purified and/or F(ab)2 second antibodies may be used if required to eliminate background.
- Seal the plate as described earlier and repeat the incubation and washings as before.
- Resuspend the cells at about 5 × 105/ml in wash buffer and analyze in the flow cytometer. The precise final cell concentration will depend upon local operating conditions and protocols.
Fluorescent-Activated Cell Sorting (FACS)
- Harvest the hES cells using trypsin:EDTA as for analysis.
- Pellet the cells and resuspend in primary antibody, diluted in medium without added azide, as determined by prior titration (100 µl per 107 cells). The primary and secondary antibodies should be sterilized using a 0.2-µm cellulose acetate filter.
- Incubate the cells with occasional shaking at 4°C for 20-30 min.
- Wash the cells by adding 10ml medium and pellet by centrifugation at 200g for 5 min; repeat this wash step once more.
- Remove supernatant and flick gently to disperse the pellet. Add 100 µl of diluted secondary antibody per 107 cells and incubate, with occasional shaking, at 4°C for 20min.
- Wash the cells two times as just described. After the final wash, resuspend the cells in medium at 107 cells/ml. Sort cells using the flow cytometer according to local protocols.
This work was supported in part by grants from the Wellcome Trust, Yorkshire Cancer Research, and the BBSRC.
References
Ackerman, S. L., Knowles, B. B., and Andrews, P. W. (1994). Gene regulation during neuronal and non-neuronal differentiation of NTERA2 human teratocarcinoma-derived stem cells. Mol. Brain Res. 25, 157-162.
Andrews, P. W. (1982). Human embryonal carcinoma cells in culture do not synthesize fibronectin until they differentiate. Int. J. Cancer 30, 567-571.
Andrews, P. W. (1984). Retinoic acid induces neuronal differentiation of a cloned human embryonal carcinoma cell line in vitro. Dev. Biol. 103, 285-293.
Andrews, P. W., Banting, G., Damjanov, I., Arnaud, D., and Avner, P. (1984a). Three monoclonal antibodies defining distinct differentiation antigens associated with different high molecular weight polypeptides on the surface of human embryonal carcinoma cells. Hybridoma 3: 347-361.
Andrews, P. W., Bronson, D. L., Benham, E, Strickland, S., and Knowles, B. B. (1980). A comparative study of eight cell lines derived from human testicular teratocarcinoma. Int. J. Cancer 26, 269-280.
Andrews, P. W., Casper, J., Damjanov, I., Duggan-Keen, M., Giwercman, A., Hata, J. I., von Keitz, A., Looijenga, L. H. J., Millán, J. L., Oosterhuis, J. W., Pera, M., Sawada, M., Schmoll, H. J., Skakkebaek, N. E., van Putten, W., and Stern, P. (1996). Comparative analysis of cell surface antigens expressed by cell lines derived from human germ cell tumors. Int. J. Cancer 66, 806-816.
Andrews, P. W., and Damjanov, I. (1994). Cell lines from human germ cell tumours. In "Atlas of Human Tumor Cell Lines" (R. J. Hay, J.-G. Park, and A. Gazdar, eds.), pp. 443-476. Academic Press, San Diego.
Andrews, P. W., Damjanov, I., Berends, J., Kumpf, S., Zappavingna, V., Mavilio, E, and Sampath, K. (1994). Inhibition of proliferation and induction of differentiation of pluripotent human embryonal carcinoma cells by osteogenic protein-1 (or bone morphogenetic protein-7). Lab. Invest. 71, 243-251.
Andrews, P. W., Damjanov, I., Simon, D., Banting, G., Carlin, C., Dracopoli, N. C., and Fogh, J. (1984b). Pluripotent embryonal carcinoma clones derived from the human teratocarcinoma cell line Tera-2: Differentiation in vivo and in vitro. Lab. Invest. 50, 147-162.
Andrews, P. W., Damjanov, I., Simon, D., and Dignazio, M. (1985). A pluripotent human stem cell clone isolated from the TERA-2 teratocarcinoma line lacks antigens SSEA-3 and SSEA-4 in vitro but expresses these antigens when grown as a xenograft tumor. Differentiation 29, 127-135.
Andrews, P. W., Goodfellow, P. N., and Bronson, D. L. (1983). Cellsurface characteristics and other markers of differentiation of human teratocarcinoma cells in culture. In "Teratocarcinoma Stem Cells" (Silver, Martin, and Strickland, eds.), pp. 579-590. Cold Spring Harbor Press, Cold Spring Harbor, NY.
Andrews, P. W., Goodfellow, P. N., Shevinsky, L. H., Bronson, D. L., and Knowles, B. B. (1982). Cell-surface antigens of a clonal human embryonal carcinoma cell line: Morphological and antigenic differentiation in culture. Int. J. Cancer 29, 523-531.
Andrews, P. W., Meyer, L. J., Bednarz, K. L., and Harris, H. (1984c). Two monoclonal antibodies recognizing determinants on human embryonal carcinoma cells react specifically with the liver isozyme of human alkaline phosphatase. Hybridoma 3, 33-39.
Andrews, P. W., Nudelman, E., Hakomori, S., and Fenderson, B. A. (1990). Different patterns of glycolipid antigens are expressed following differentiation of TERA-2 human embryonal carcinoma cells induced by retinoic acid, hexamethylene bisacetamide HMBA or bromodeoxyuridine BUdR. Differentiation 43, 131-138.
Andrews, P. W., Trinchieri, G., Perussia, B., and Baglioni, C. (1987). Induction of class 1 major histocompatibility complex antigens in human teratocarcinoma cells by interferon without induction of differentiation, growth inhibition or resistance to viral infection. Cancer Res. 47, 740-746.
Badcock, G., Pigott, C., Goepel, J., and Andrews, P. W. (1999). The human embryonal carcinoma marker antigen TRA-1-60 is a sialylated keratan sulfate proteoglycan. Cancer Res. 59, 4715-4719.
Bernstine, E. G., Hooper, M. L., Grandchamp, S., and Ephrussi, B. (1973). Alkaline phosphatase activity in mouse teratoma. Proc. Natl. Acad. Sci. USA. 70, 3899-3903.
Bottero, L., Simeone, A., Arcioni, L., Acampora, D., Andrews, P. W., Boncinelli, E., and Mavilio, E (1991). Differential activation of homeobox genes by retinoic acid in human embryonal carcinoma cells. In "Recent Results in Cancer Research" (J. W. Oosterhuis, H. Walt, and I. Damjanov, eds.), Vol. 123, pp. 133-143. Springer- Verlag, New York.
Damjanov, I., and Andrews, P. W. (1983). Ultrastructural differentiation of a clonal human embryonal carcinoma cell line in vitro. Cancer Res. 43, 2190-2198.
Draper, J. S., Pigott, C., Thomson, J. A., and Andrews, P. W. (2002). Surface antigens of human embryonic stem cells: Changes upon differentiation in culture. J. Anat. 200, 249-258.
Duran, C., Talley, P. J., Walsh, J., Pigott, C., Morton, I., and Andrews, P. W. (2001). Hybrids of pluripotent and nullipotent human embryonal carcinoma cells: Partial retention of a pluripotent phenotype. Int. J. Cancer 93, 324-332.
Eisenbarth, G. S., Walsh, E S., and Nirenberg, M. (1979). Monoclonal antibody to a plasma membrane antigen of neurons. Proc. Natl. Acad. Sci. USA 76, 4913-4917.
Fenderson, B. A., Andrews, P. W., Nudelman, E., Clausen, H., and Hakomori, S. (1987). Glycolipid core structure switching from globo- to lacto- and ganglio-series during retinoic acid-induced differentiation of TERA-2-derived human embryonal carcinoma cells. Dev. Biol. 122, 21-34.
Fogh, J., and Trempe, G. (1975). New human tumor cell lines. In "'Human Tumor Cells in Vitro" (J. Fogh, ed.), pp. 115-159. Plenum Press, New York.
Gönczö1, E., Andrews, P. W., and Plotkin, S. A. (1984). Cytomegalovirus replicates in differentiated but not undifferentiated human embryonal carcinoma cells. Science 224, 159-161.
Gooi, H. C., Feizi, T., Kapadia, A., Knowles, B. B., Solter, D., and Evans, M. J. (1981). Stage specific embryonic antigen involves α1 → 3 fucosylated type 2 blood group chains. Nature 292, 156-158.
Hirka, G., Prakesh, K., Kawashima, H., Plotkin, S. A., Andrews, P. W., and Gönczö1, E. (1991). Differentiation of human embryonal carcinoma cells induces human immunodeficiency virus permissiveness which is stimulated by human cytomegalovirus coinfection. J. Virol. 65, 2732-2735.
Kannagi, R., Cochran, N. A., Ishigami, E, Hakomori, S.-I., Andrews, P. W., Knowles, B. B., and Solter, D. (1983). Stage-specific embryonic antigens SSEA3 and -4 are epitopes of a unique globo-series ganglioside isolated from human teratocarcinoma cells. EMBO J. 2, 2355-2361.
Kannagi, R., Nudelman, E., Levery, S. B., and Hakomori, S. (1982). A series of human erythrocyte glycosphingolipids reacting to the monoclonal antibody directed to a developmentally regulated antigen, SSEA1. J. Biol. Chem. 257, 14865-14874.
Kohler, G., and Milstein, C. (1975). Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 256, 495-497.
LaFemina, R., and Hayward, G. S., (1986). Constitutive and retinoic acid-inducible expression of cytomegalovirus immediate-early genes in human teratocarcinoma cells. J. Virol. 58, 434-440.
Lee, V. M-Y., and Andrews, P. W. (1986). Differentiation of NTERA- 2 clonal human embryonal carcinoma cells into neurons involves the induction of all three neurofilament proteins. J. Neurosci. 6, 514-521.
Martin, G. R., and Evans, M. J. (1974). The morphology and growth of a pluripotent teratocarcinomas cell line and its derivatives in tissue culture. Cell 2, 163-172.
Matthaei, K., Andrews, P. W., and Bronson, D. L. (1983). Retinoic acid fails to induce differentiation in human teratocarcinoma cell lines that express high levels of cellular receptor protein. Exp. Cell Res. 143, 471-474.
Nelson, J. A., Reynolds-Kohler, C., and Smith, B. A., (1987). Negative and positive regulation by a short segment in the 5'-flanking region of the human cytomegalovirus major immediate-early gene. Mol. Cell. Biol. 7, 4125-4129.
Pera, M. E, Blasco-Lafita, M. J., Cooper, S., Mason, M., Mills, J., and Monaghan, P. (1988). Analysis of cell-differentiation lineage in human teratomas using new monoclonal antibodies to cytostructural antigens of embryonal carcinoma cells. Differentiation 39, 139-149.
Pleasure, S. J., and Lee, V. M. Y. (1993). NTERA-2 cells a human cell line which displays characteristics expected of a human committed neuronal progenitor cell. J. Neurosci. Res. 35, 585-602.
Pleasure, S. J., Page, C., and Lee, V. M.-Y. (1992). Pure, post-mitotic, polarized human neurons derived from Ntera2 cells provide a system for expressing exogenous proteins in terminally differentiated neurons. J. Neurosci. 12, 1802-1815.
Przyborski, S. A., Morton, I. E., Wood, A., and Andrews, P. W. (2000). Developmental regulation of neurogenesis in the pluripotent human embryonal carcinoma cell line NTERA-2. Eur. J. Neurosci. 12, 3521-3528.
Rendt, J., Erulkar, S., and Andrews, P. W. (1989). Presumptive neurons derived by differentiation of a human embryonal carcinoma cell line exhibit tetrodotoxin-sensitive sodium currents and the capacity for regenerative responses. Exp. Cell Res. 180, 580-584.
Reubinoff, B. E., Pera, M. F., Fong, C. Y., Trounson, A., and Bongso, A. (2000). Embryonic stem cell lines from human blastocysts: somatic differentiation in vitro. Nature Biotechnol 18, 399-404.
Simeone, A., Acampora, D., Arcioni, L., Andrews, P. W., Boncinelli, E., and Mavilio, F. (1990). Sequential activation of human HOX2 homeobox genes by retinoic acid in human embryonal carcinoma cells. Nature 346, 763-766.
Solter, D., and Knowles, B. B. (1978). Monoclonal antibody defining a stage-specific mouse embryonic antigen SSEA1. Proc. Natl. Acad. Sci. USA 75, 5565-5569.
Thompson, S., Stern, P. L., Webb., M., Walsh, E S., Engstr6m, W., Evans, E. P., Shi, W. K., Hopkins, B., and Graham, C. E (1984). Cloned human teratoma cells differentiate into neuron-like cells and other cell types in retinoic acid. J. Cell Sci. 72, 37-64.
Thomson, J. A., Itskovitz-Eldor, J., Shapiro, S. S., Waknitz, M. A., Swiergiel, J. J., Marshall, V. S., and Jones, J. M. (1998). Embryonic stem cell lines derived from human blastocysts. Science 282, 1145-1147.
Thurin, J., Herlyn, M., Hindsgaul, O., Stromberg, N., Karlsson, K. A., Elder, D., Steplewski, Z., and Koprowski, H. (1985). Proton NMR and fast-atom bombardment mass spectrometry analysis of the melanoma-associated ganglioside 9-O-acetyl-GD3. J. Biol. Chem. 260, 14556-14563.
Support our developers




