Differentiation of Pancreatic Cells into Hepatocytes
The phenomenon of transdifferentiation is defined as the conversion of one differentiated cell type to another (Tosh and Slack, 2002). Generally, cells that have the potential to transdifferentiate arise from adjacent regions in the developing embryo. Therefore, transdifferentiation between adult cells probably reflects their close developmental relationship. Numerous examples of transdifferentiation have been described in literature (Eguchi and Kodama, 1993). Two examples are the transdifferentiation of pancreas to liver (reviewed in Tosh and Slack, 2002; Shen et al., 2003) and the reverse, liver to pancreas conversion (Horb et al., 2003). The liver and pancreas exhibit a close developmental relationship, as they arise from the same region of the endoderm (Wells and Melton, 1999). We have developed two different in vitro approaches for inducing the transdifferentiation of pancreatic cells to hepatocytes. The conversion of pancreatic cells to hepatocytes can be induced by culture of either the pancreatic cell line AR42J (Longnecker et al., 1979; Christophe, 1994) or the mouse embryonic pancreas (Percival and Slack, 1999). The first system, AR42J cells, is a cell line originally isolated from a carcinoma of an azaserine-treated rat (Longnecker et al., 1979); although a single cell type, they are considered to be amphicrine in nature, i.e., they possess both exocrine and neuroendocrine properties (Christophe, 1994). The expression of the exocrine enzyme amylase can be enhanced by short-term culture with 10nM dexamethasone (Logsdon et al., 1985). The advantage of the second system for studying transdifferentiation, the cultured embryonic pancreas system, is that it is more physiological than the AR42J cell line, which has been around for more than 20 years (Shen et al., 2000). In addition, as the dorsal pancreatic organ grows as a flattened, branched structure, it is suitable for whole mount immunostaining. As well as being of interest to individuals who plan to investigate the transdifferentiation of pancreas to liver, the system is also relevant to those working on normal pancreas development. This article describes the use of AR42J cells and mouse embryonic pancreas as models for the transdifferentiation of pancreas to liver.
Several protocols have been produced for inducing the in vivo appearance of hepatocytes in the pancreas. For example, administration of a methionine-deficient diet and exposure to a carcinogen (Scarpelli and Rao, 1981) can induce hepatocytes in the pancreas of hamsters. However, one of the most efficient means of converting pancreas to liver is to feed rats a copper-deficient diet in combination with a copper chelator, Trien (Rao et al., 1988). After 7-9 weeks of copper deficiency, the animals are returned to their normal diet and hepatocytes begin to appear soon afterwards. Hepatocytes in the pancreas express a range of liver-specific proteins, e.g., albumin, and are able to respond to xenobiotics (Rao et al., 1982, 1988).
One drawback to in vivo studies is that it is difficult to study individual changes at the cellular or molecular levels. An alternative approach is to use in vitro models, e.g., AR42J cells. The hepatocytes that are induced to differentiate from pancreatic AR42J Cells express many of the proteins that are found in normal liver, e.g., albumin, glucose-6-phosphatase, transferrin, transthyretin, and proteins involved in detoxification (e.g., UDP-glucuronosyltransferases, CYP family) (Shen et al., 2000; Tosh et al., 2002). This system offers the ability to generate hepatocytes that express a whole range of liver proteins while at the same time avoiding the necessity to isolate hepatocytes from in vivo. It also permits the opportunity to study factors for inducing the conversion of one cell type to another. The in vitro culture of mouse embryonic pancreas is particularly suitable for whole mount immunostaining. This in turn provides a three-dimensional visualisation of the cell arrangements (Percival and Slack, 1999; Horb and Slack, 2000). The current system offers a relatively simple approach and depends on the presence of a substrate (in this case fibronectin), orientation of the cut pancreas, and a serum-rich medium.
Both AR42J cells and embryonic pancreas can be induced to transdifferentiate to hepatocytes by exposure to the glucocorticoid dexamethasone. We find 1 µM dexamethasone to be sufficient. To unambiguously demonstrate the conversion of pancreatic cells to hepatocytes, a number of criteria have to be fulfilled. These include (1) the phenotypic characterisation of the parent cells (i.e., pancreatic cells) and the descendants (the liver cells) and (2) the lineage relationship between the ancestor and the descendant. Characterization of the phenotypes can be morphological and biochemical and/or molecular. In the case of AR42J cells, they exhibit an exocrine phenotype. Therefore the cells can be characterised with markers of digestive enzymes, e.g., amylase. Because the embryonic pancreas contains both exocrine and endocrine cell types, it is possible, with the correct combination of antibodies, to immunostain for at least three cell types (exocrine cells, glucagon-secreting α cells, and insulinsecreting β cells). In contrast, hepatocytes exhibit a vast array of proteins, including albumin, transferrin, and transthyretin so it is easy to determine the expression of the descendant. The second criterion (to demonstrate the ancestor-descendant relationship) can be satisfied by using the Cre-lox system in vivo (Herrera, 2000) or by lineage labelling in vitro, e.g., using green fluorescent protein (GFP) (Shen et al., 2000).
Dexamethasone can be replaced by the naturally occurring glucocorticoid cortisol to induce the conversion of AR42J cells to hepatocytes. To determine whether the effect of the glucocorticoid is specific, the cells can be exposed to RU486, the glucocorticoid receptor antagonist, prior to the addition of dexamethasone or cortisol. Furthermore, the number of AR42J cells that will transdifferentiate can be enhanced by the culture of dexamethasone in combination with oncostatin M. Oncostatin M is a member of the IL-6 family of interleukins and has been shown to enhance the maturation of embryonic liver (Kamiya et al., 1999).
Dexamethasone (D1756) and cortisol are from Sigma Chemical Co (St. Louis, MO). RU-486 (Mifepristone) is from Biomol Research Laboratories, Inc. (Plymouth, PA). Recombinant human oncostatin M is from R&D System Inc. (Minneapolis, MN). Dulbecco's minimum essential medium (DMEM; D5546), basal medium Eagle (BME, B1522), minimum essential medium Eagle (MEM Eagle, M5775), penicillin-streptomycin solution (10,000U/ml/10-mg/ml stock, P4333), and L-glutamine (G7153) are from Sigma (Poole, Dorset, UK). Trypsin-EDTA solution (25300- 054), gentamycin (15710-049), and fetal bovine serum (FBS) (10106-169) are all from Invitrogen (Paisley, UK). Tissue culture 35-mm plastic dishes are from Fischer and 22 × 22-mm (MIC 3114) glass coverslips are from Scientific Laboratory Supplies (Nottingham, UK). Blocking reagent (1 096 176) is from Roche (Welwyn Garden City, UK).
Phosphate-buffered saline (PBS) tablets are supplied by Oxoid Ltd. (Basingstoke, UK). MilliQ-filtered H2O is sterilised by autoclaving. Dissecting instruments, including small scissors, large scissors, tungsten wire needle (Goodfellow Metals, Cambridge, UK) in a glass capillary tube, and two pairs of forceps (Dumont No. 5, Sigma F6521), are required.
A. Cell Lines and Culture Conditions
Transdifferentiation of AR42J Cells to Hepatocytes
AR42J cells can be obtained as a frozen aliquot or growing culture from the ECACC (93100618) or ATCC (CRL-1492). AR42J-B13 cells (kindly provided by Dr. Itaru Kojima, Japan) are a subclone of the parent line AR42J. The subclone was isolated on the basis of an increased tendency to convert to β cells (Mashima et al., 1996). Either cell type can be induced to transdifferentiate to hepatocytes, the difference being that the AR42J-B13 subclone transdifferentiates more readily than the parent cell line (Shen et al., 2000). Cells are maintained in Dulbecco's modified Eagle's medium containing penicillin, streptomycin, and 10% fetal bovine serum. Dex (1 µM) is added as a solution in ethanol, and medium is changed every 1-2 days. RU486 is added at a concentration of 2.5 µM, with the treatment commencing 1 h before addition of the Dex. Oncostatin M is added as a solution in phosphatebuffered saline containing 0.1% bovine serum albumin at a final concentration of 10ng/ml together with 1 µM Dex.
B. Other Procedures
Immunofluorescence Analysis and Antisera
- For immunofluorescent staining, culture AR42JB13 cells on noncoated glass coverslips, rinse with PBS, and fix with 4% paraformaldehyde in PBS for 30rain. Immunostain the cells on a coverslip as described later and mount on a microscope slide in a suitable mounting media such as gelvatol.
- Permeabilise with 0.1% (v/v) Triton X-100 in PBS for 30 rain.
- Incubate the coverslips in 2% blocking buffer (Roche) and 0.1% (v/v) Triton X-100, and then incubate sequentially with primary and secondary antibodies. Dilute and obtain the antibodies as follows: rabbit polyclonal antiamylase (A8273; 1/300) and rabbit polyclonal antialbumin (1/500; A0433), both from Sigma Chemical Co., and rabbit polyclonal antitransferrin (A0061, 1/200) and rabbit polyclonal antitransthyretin (A0062, 1/100), both from DAKO (Ely, Cambridge). The goat polyclonal antirabbit IgG FITC conjugate (FI-1000; 1/150) is from Vector Laboratories (Fig. 1).
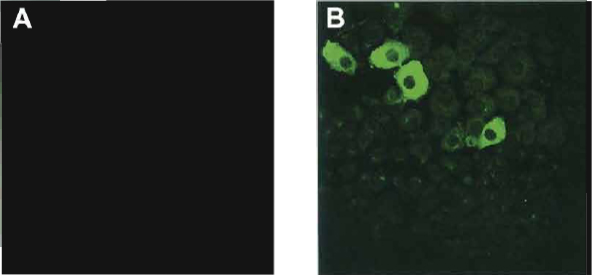 |
| FIGURE 1 Immunostaining for albumin in control (A) and dexamethasone-treated AR42J-B13 cells (B). |
Gelvatol Medium
- Produce gelvatol medium by dissolving 20g of polyvinyl alcohol in 80ml of 10mM Tris, pH 8.6, along with 0.2% NaN3.
- In a separate tube, mix 3g of n-propyl gallate and 50ml glycerol until clear. This will take 2-3 days at room temperature with rotating.
- When both solutions are dissolved, mix the two. Occasionally lumps are present, which can be removed by centrifugation at 2500rpm for 15 min.
- Pour off the supernatant into a new tube, leaving the lumps at the bottom of the old tube.
- This tube can now be wrapped in foil and stored at 4°C.
C. Isolation of Mouse Embryonic Pancreas
This procedure is modified from Percival and Slack (1999) and from Horb and Slack (2000).
Embryonic Culture of Mouse Pancreatic Buds
Isolate dorsal pancreatic buds from 11.5-day mouse embryos as described later. Following isolation, dissect the pancreatic buds out in minimum essential medium with Hank's salts (Hank's medium) containing 10% FBS, 2mM glutamine, and 50µg/ml gentamycin. Culture the buds on fibronectin-coated coverslips in medium containing basal medium Eagle with Earle's salts (Earle's medium), 2 mM glutamine, 50 gg/ml gentamycin, and 20% FBS. Change the medium daily for up to 6 days.
Fibronectin comes as a lyophilised powder (Invitrogen, Cat. No. 33010-018).
- Add 1 ml of sterilised water to a 1-mg vial of fibronectin and dissolve (1 mg/ml final concentration).
- Aliquot 50µl into 1-ml Eppendorfs and freeze at -20°C.
- Prepare glass coverslips by baking for at least 3 h at 180°C.
- Prior to adding fibronectin, coat the coverslips with 2% 3-aminoporpyltriethoxysilane (APTS) to enhance attachment of the bud to the coverslip.
- To coat the coverslip with fibronectin, take one tube of the frozen 50 µl fibronectin and add 950 µl of sterilised water (make sure it is well dissolved).
- In the tissue culture hood, pipette 50µl of solution onto a sterilised coverslip that has been placed in a 35-mm culture dish. The final concentration of fibronectin is 50µg/ml.
- Allow the fibronectin to dry on the coverslips. This takes 2-3 h.
Preparation of Embryos
Mouse embryos are generated by timed matings. The day of the vaginal plug is taken as 0.5. For the purpose of the present study, we use 11.5-day embryos.
- Kill mouse by cervical dislocation.
- Test reflexes generally by gripping the paw. If reflexes are absent, then proceed.
- Soak the fur in 70% ethanol. This is generally a source of infection.
- Using blunt forceps and scissors, cut open the abdomen (first layer) by a small incision and then tear this back with the fingers and expose the peritoneal sack.
- Cut the layer with the sharp scissors and forceps and expose one of the uterine horns by displacing the abdominal contents and fatty tissue to the side.
- Find the furthest embryo from the base of the uterus in either horn and remove the whole string of embryos into a 90-mm petri dish containing ice-cold PBS.
- Separate the embryos from each other using blunt forceps and scissors; alternatively, simply hold the embryo and uterus with blunt forceps in one hand and squeeze gently until the embryo pops out of the uterus. Place embryos in a clean petri dish of PBS.
- Dissect the embryos from the sacs and remove their heads. Place in a new petri dish containing Hank's medium.
- Dissect open the embryo and locate the stomach. At the lower end, just as the stomach empties into the intestine, there is a tissue lying over the surface of the organ. This is the dorsal embryonic bud. Separate the stomach (along with the dorsal pancreas) from the intestine and liver using the needle as a knife. Always remember to cut away from the tissue that you require to avoid damaging the delicate tissue. Finally, with the needle, prise the pancreas away from the stomach and place in a fresh dish of Hank's medium.
- To culture the pancreatic bud, first place a cloning ring on top of the fibronectin-coated region of the coverslip. Fibronectin is visible as a dried "ring" on the coverslip so it is easy to locate. Add 2.5ml of Earle's culture medium. Initially add some medium dropwise to the cloning ring but avoid bubbles, as this makes it difficult for the pancreas to settle and attach to the substratum.
- For each pancreas culture, suck up tissue into a pipette and lower onto medium in the cloning ring. Although two to three pancreatic buds can be cultured within a single cloning ring, they sometimes grow and overlap so it is best to culture only one per dish.
- Centre the pancreas with the tungsten needle and make sure that the cut surface of the pancreas bud is face down. It is important that the culture is placed in the centre, as occasionally the cloning ring can move slightly and could crush the bud, especially if it is too close to the edge of the cloning ring. Also, ensuring that the bud is placed cut surface down will increase the chances of the bud sticking to the fibronectin matrix.
- Place the dish in the incubator at 37°C and culture overnight.
- The following day, check the cultures under an inverted microscope. Generally, allow 24h before changing the medium. Remove the cloning rings from the coverslips with forceps and then change the medium to fresh medium. Repeat medium changes every 1-2 days (Fig. 2).
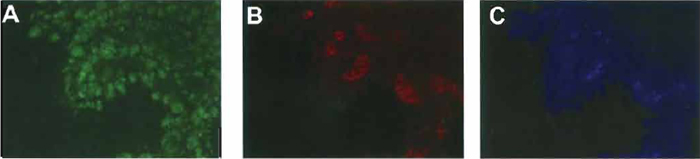 |
| FIGURE 2 Immunostaining for amylase, insulin, and glucagon in normal pancreatic buds. Pancreatic buds were cultured for 7 days and then immunostained for amylase (A), insulin, (B), and glucagon (C). |
Induction of Transdifferentiation
To induce transdifferentiation of pancreatic cells to hepatocytes, add 1 µM dexamethasone. This should be added at 1- to 2-day intervals from the 1 mM stock that is kept at-20°C. Dexamethasone can be added to the medium prior to pipetting onto the dishes or directly to the dish. Add an equivalent volume of ethanol to control dishes.
The embryonic buds can be analysed by immunofluorescent analysis, but the fixation and permeabilisation conditions are different from those for AR42J cells.
D. Immunofluorescence Analysis of Embryonic Pancreas
- For immunofluorescent staining, fix the pancreata in MEMFA (10% formaldehyde, 0.1M Mops, pH 7.4, 2 mM, EGTA, 1 mM MgSO4) for 30-45 min at room temperature.
- Wash in PBS; they can be stored in PBS at 4°C for up to a few days.
- Prior to immunostaining, treat the cultures with 1% Triton X-100 in PBS to permeabilise and then block in 2% blocking buffer (Roche), which contains 0.1% Triton X-100.
- The buds can then be incubated sequentially with primary and secondary antibodies. We can perform triple labelling for the detection of amylase (FITC), insulin (TRITC), and glucagon (AMCA). Dilute the primary antibodies in blocking buffer and apply to the coverslip overnight at 4°C. Dilute and obtain the antibodies as follows: rabbit polyclonal antiamylase (1/300, Sigma A8273), guinea pig polyclonal antiinsulin (1/300, Sigma, I6163), mouse monoclonal antiglucagon (1:100, Sigma G2654), goat polyclonal antirabbit IgG FITC conjugate (1/100; Vector Laboratories FI-1000), rabbit polyclonal antiguinea pig IgG TRITC conjugate (1/300; Sigma T7153), and horse antimouse IgG AMCA conjugate (1/100; Vector Laboratories CI-2000). The following day, wash the coverslips three times (15 min each) and apply the secondary antibody and leave on for 3 h. On the last day (day 4), wash the coverslip again (three times 15min each) and mount the coverslip in an appropriate medium, e.g., gelvatol or mobiol. View specimens under a fluorescent microscope. After induction of transdifferentiation with dexamethasone, the hepatocytes can be detected in buds by using the liver antibodies described in Section III,B.
For confocal imaging, we use a Zeiss LSM 510 confocal system on an inverted Zeiss fluorescent microscope fitted with a ×40/NA 1.30 or ×63/NA 1.40 oil immersion objective (Zeiss, Welwyn Garden City, UK). Alternatively, we use a Leica DMRB microscope fitted with a digital camera. Generally, when two or more antibodies are visualized through different fluorescent channels in the same specimen, the initial black-andwhite CCD images are coloured and then recombined to form a single multicoloured image.
IV. COMMENTS
The differentiation of pancreatic AR42J cells to hepatocytes results in a marked morphological change. The cells become flattened and enlarged. These changes occur with 5 days of dexamethasone treatment. It is therefore possible to check the differentiation by noting the number and degree of flattening of cells.
V. PITFALLS
- For the best results with the embryonic pancreatic cultures, ensure that the bud is placed cut surface down and in the centre of the cloning ring. If not, there is a chance that the cloning ring will move when the incubator door is closed, squashing the bud.
- When immunostaining for liver proteins, do not be tempted to use serum as a blocking agent. Although good for conventional immunostaining, it is not suitable for some of the liver markers, which are serum proteins normally secreted from the organ, e.g., albumin.
- Prior to inducing transdifferentiation, inoculate the AR42J cells at low density (e.g., we routinely inoculate at 10-15%). These cells have a high rate of cell division and although dexamethasone inhibits cell division, you may end up with too confluent a dish for optimal immunofluorescence.
This work was supported by the Medical Research Council.
References
Christophe, J. (1994). Pancreatic tumoral cell-line AR42J. An amphicrine model. Am. J. Physiol. 266, G963-G971.
Eguchi, G., and Kodama, R. (1993). Transdifferentiation. Curr. Opin. Cell Biol. 5, 1023-1028.
Herrera, P. L. (2000). Adult insulin and glucagon cells differentiate from two independent cell lineages. Development 127, 2317-2322.
Horb, U D., and Slack, J. M. W. (2000). Role of cell division in branching morphogenesis and differentiation of the embryonic pancreas. Int. J. Dev. Biol. 44, 791-796.
Horb, M. E., Shen, C.-N., Tosh, D., and Slack, J. M. W. (2003). Experimental conversion of liver to pancreas. Curr. Biol. 13, 105-115.
Logsdon, C. D., Moessner, J., William, J. A., and Goldfine, I. D. (1985). Glucocorticoids increase amylase mRNA levels, secretory organelles, and secretion in pancreatic acinar AR42J cells. J. Cell Biol. 100, 1200-1208.
Longnecker, D. S., Lilja, H. S., French, J. I., Kuhlmann, E., and Noll, W. (1979). Transplantation of azeserine-induced carcinomas of pancreas in rats. Cancer Lett. 7, 197-202.
Kamiya, A., et al. (1999). Fetal liver development requires a paracrine action of oncostatin M through gp130 signal transducer. EMBO J. 18, 2127-2136.
Mashima, H., Shibata, H., Mine, T., and Kojima, I. (1996). Formation of insulin-producing cells from pancreatic acinar AR42J cells by hepatocyte growth factor. Endocrinology 137, 3969-3976.
Rao, M. S., Reddy, M. K., Reddy, J. K., and Scarpelli, D. G. (1982). Response of chemically induced hepatocyte-like cells in hamster pancreas to methyl clofenapate, a peroxisomal proliferator. J. Cell Biol. 95, 50-56.
Rao, M. S., et al. (1988). Almost total conversion of pancreas to liver in the adult rat: A reliable model to study transdifferentiation. Biochem. Biophys. Res. Commun. 156, 131-136.
Scarpelli, D. G., and Rao, M. S. (1981). Differentiation of regenerating pancreatic cells into hepatocyte-like cells. Proc. Natl. Acad. Sci. USA 78, 2577-2581.
Shen, C.-N., Horb, M. E., Slack, J. M. W., and Tosh, D. (2003). Transdifferentiation of pancreas to liver. Mech. Dev. 120, 107-116.
Shen, C.-N., Slack, J. M. W., and Tosh, D. (2000). Molecular basis of transdifferentiation of pancreas to liver. Nature Cell Biol. 2, 879-887.
Tosh, D., Shen, C.-N., and Slack, J. M. W. (2002). Differentiated properties of hepatocytes induced from pancreatic cells. Hepatology 36, 534-543.
Tosh, D., and Slack, J. M. W. (2002). How cells change their phenotype. Nature Rev. Mol. Cell Biol. 3, 187-194.
Wells, J. M., and Melton, D. A. (1999). Vertebrate endoderm development. Annu. Rev. Cell Dev. Biol. 15, 393-410.
Support our developers




