Biosynthesis of Allyl and Propenyl Phenols and Related Phenylpropanoid Moieties
Besides lignin, specialized plant metabolism can utilize monolignols in the formation
of lignans (phenylpropanoid dimers) and, as recently elucidated, allyl and
propenyl phenols. Allylphenols differ from propenylphenols in their side-chain
double bond position, with the former having terminal (C-8–C-9) desaturation
and the latter having the chemically more stable internal (C-7–C-8) double bond.
Several biochemical hypotheses had been created to explain their distinctive lack
of a C-9 oxygenated functionality (Canonica
et al., 1971; Klischies
et al., 1975;
Manitto
et al., 1974a,b), but experimental support for any was lacking until
recently.
Our interest in the allyl/propenyl phenols first began with studies directed at
elucidating the biochemical pathway(s) to the lignan nordihydroguaiaretic acid
(
43, NDGA, Fig. 13.10) and its congeners (Cho
et al., 2003; Moinuddin
et al., 2003),
these being abundant metabolites in the creosote bush (Larrea tridentata). These
substances are of increasing interest due to their potent biological and (potential)
medicinal applications. For example, the NDGA derivative 44 is apparently
proceeding smoothly through National Institutes of Health trials as an effective
chemotherapeutic treatment for the usually hard to treat (refractory) brain
and central nervous system tumors (Chang
et al., 2004)
(see also http://www.
clinicaltrials.gov/ct/show/NCT00404248, http://www.clinicaltrials.gov/ct/show/
NCT00259818 and
http://www.cancer.gov/search/viewclinicaltrials.aspx?cdrid=
455645). Interestingly, the creosote bush has long been part of traditional Native
American Indian medicine, being used to treat more than 50 diseases, most
commonly those of renal and gynecologic origins (Arteaga
et al., 2005). These
lignans, however, lack oxygenated carbon 9,90 functionalities that are present in
most lignan classes [e.g., podophyllotoxin (
45), secoisolariciresinol (
46),
Fig. 13.10], as well as in the polymeric lignins and monomeric phenylpropanoids
(e.g., monolignols 19–23) in the vast majority of plant species. Based on previous
radiolabeling/stable isotope labeling studies (Moinuddin
et al., 2003), it was
presumed that the unusual ‘‘loss’’ of the oxygenated functionality occurred at the monomer stage; that is, allyl and/or propenyl phenols could be serving as the
precursors/substrates for
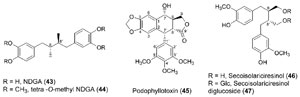 |
| FIGURE 13.10 Lignans commonly used in
medicinal applications, particularly for cancer
treatment/prevention. |
dimerization to form these less common lignans. In this
regard, the biosynthetic pathways to the allyl/propenyl phenols had not been
elucidated in any organism and, in particular, the precise precursor (substrate)
undergoing deoxygenation represented both a long-standing question and a
biochemical mystery (Canonica
et al., 1971; Klischies
et al., 1975; Manitto
et al.,
1974a,b; Senanayake
et al., 1977).
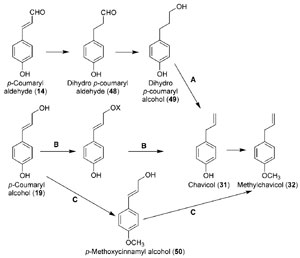 |
| FIGURE 13.11 Hypothetical biosynthetic
pathways from p-coumaryl alcohol (19) to
chavicol (31)
and methylchavicol (32), of which
pathway B was demonstrated to occur. Source:
Vassão et al.
(2006b). |
Basil (
Ocimum basilicum, Thai variety) was used as a suitable study system
since it accumulates the simplest allylphenol, methylchavicol (
32); based on
various radiolabeling studies, it was shown that the latter was derived from the
corresponding monolignol,
p-coumaryl alcohol (
19) (Vassão
et al., 2006b). Three
potential mechanisms for the conversion of 19 into 32 included reduction of the
monolignol side-chain (i.e., saturation) followed by dehydration (Fig. 13.11A);
methylation of the phenolic moiety preceding further side-chain modification
(Fig. 13.11C); and/or activation of the terminal (C-9) oxygenated functionality
prior to side-chain double bond reduction (Fig. 13.11B). Pathways A and C
(Fig. 13.11) were eliminated since no experimental evidence in support of either
route was obtained.
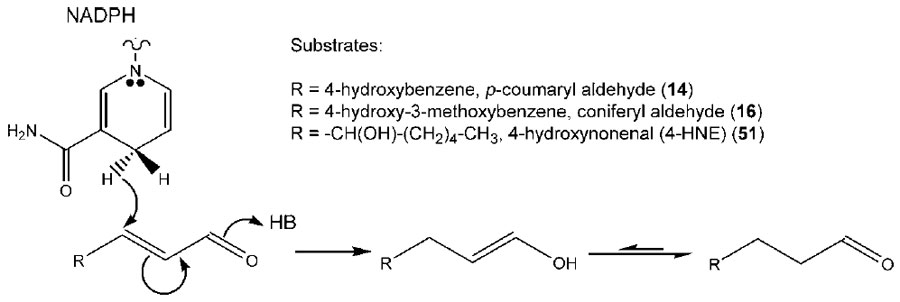
Interestingly, however, a double-bond reductase was discovered and characterized,
which utilized
p-coumaryl aldehyde (
14) as the preferred substrate to
afford the corresponding side-chain reduced aldehyde (
48, Fig. 13.11) (Kasahara
et al., 2004, 2006; Youn
et al., 2006b). This alkenal reductase activity was the first to
be reported in the phenylpropanoid pathway, with the corresponding
enzymes
isolated from
A. thaliana (AtDBR) and
Pinus taeda (PtPPDBR) also being homologous
to a terpenoid double-bond reductase (pulegone reductase, PulR) from
Mentha piperita and mammalian alkenal reductases as well (Fig. 13.12). AtDBR
and PtPPDBR catalyze the NADPH-
 |
| FIGURE 13.12 Alignment of double-bond
reductases from Pinus taeda (PtPPDBR),
Arabidopsis thaliana (AtDBR1), and homologues
from Mentha
piperita (pulegone reductase, PulR),
rat (Rattus norvegicus, AOR), guinea pig (Cavia
porcellus, 12-HD/PGR), and mouse (Mus
musculus, 1VJ1). The
nucleotide-binding domain
is indicated by the dotted line, with conserved
AXXGXXG motif in red. Conserved catalytic
Tyr residues (Y260 for AtDBR1)
are highlighted
in light blue, and secondary structural elements
are indicated in colored bars (blue for α-helices
and orange for β-strands). Source:
Redrawn from
Youn et al. (2006b). |
dependent reduction of
p-coumaryl (
14) and
coniferyl (
16) aldehydes to the corresponding dihydroaldehydes, (Fig. 13.13) and
AtDBR has also been shown to catalyze the reduction of 4-hydroxynonenal (4-
HNE,
51), a pro-apoptotic lipid peroxidation product, to 4-hydroxynonanal
(Kasahara
et al., 2006; Youn
et al., 2006b). Based on substrate versatility studies
and an X-ray crystal structure for AtDBR, a concerted mechanism involving an
enol intermediate was proposed for these zinc-independent alkenal reductases
(Youn
et al., 2006b). While the corresponding dihydroalcohol product 49 is a wellknown
plant defense metabolite (Kraus and Spiteller, 1997), it was not, however,
converted in basil into either chavicol (
31) and/or
p-anol (
37) (Vassão
et al., 2006b).
Accordingly, it was not considered as being involved in allyl/propenyl phenol
biosynthesis.
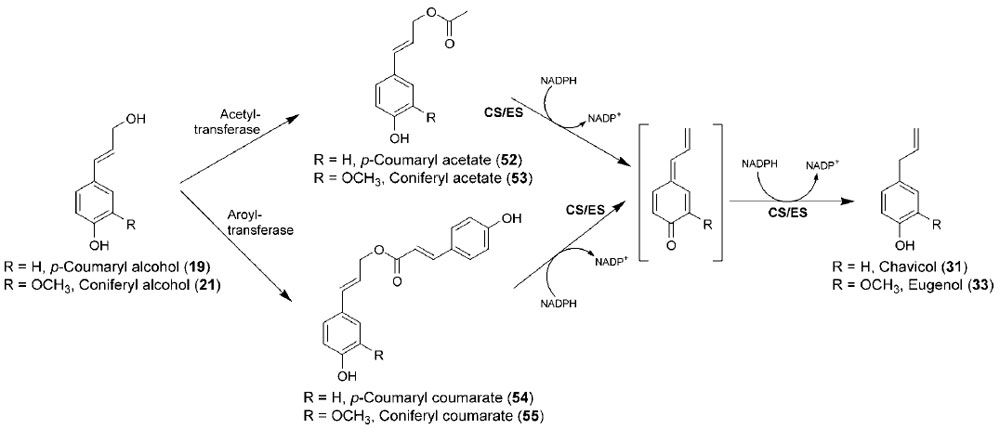
Instead, a quite novel metabolic process converting monolignols [such as
p-coumaryl (
19) and coniferyl (
21) alcohols] into allyl/propenyl phenols [chavicol
(
31) and eugenol (
33), respectively] was discovered (Fig. 13.14), with two enzymes
being implicated in their formation
in planta. The first step is activation of the
monolignol side-chain alcohol by conjugation to an activated acid (acyl-CoA),
resulting in formation of a monolignol ester. This modification results, in
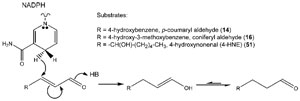 |
| FIGURE 13.13 Possible enzymatic mechanism
for AtDBR-mediated conversion of p-coumaryl
(14) and coniferyl (16) aldehydes and 4-HNE
(51) into their corresponding dihydroaldehyde
derivatives. Source: Redrawn from Youn et al.
(2006b). |
energetic
terms, in formation of a more facile leaving group (carboxylate ester), which is
more readily displaced by an incoming reducing hydride, for example, in the form
of NAD(P)H. Indeed, such coniferyl alcohol acyl transferases have been recently
characterized in basil (
O. basilicum) (Harrison and Gang, 2006) and petunia
(
Petunia hybrida) (Dexter
et al., 2007), utilizing acetyl-CoA and coniferyl alcohol
(
21) to afford coniferyl acetate (
53), and it is anticipated that substrate-versatile
acyltransferases may be able to utilize different monolignols and acyl/aroyl-CoA
cofactors to generate different esters. One such ester,
p-coumaryl coumarate (
54),
had been previously shown to serve as substrate for enzyme preparations from
Asparagus officinalis (Suzuki
et al., 2002) and
Cryptomeria japonica (Suzuki
et al.,
2004), generating the
nor-lignans (Z)- and
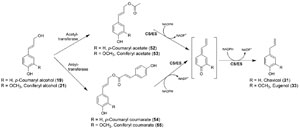 |
| FIGURE 13.14 Biosynthetic pathway to chavicol
(31) and eugenol (33) from the corresponding
monolignols, p-coumaryl (19) and coniferyl (21)
alcohols. CS, chavicol synthase; ES, eugenol
synthase. |
(E)-hinokiresinol (56/57; Fig. 13.15A)
respectively. Although the proteins responsible for the latter conversions remain
to be fully characterized and/or described, potential mechanisms whereby the
departing carboxylate (as CO
2) facilitates formation of the final C8–C70 bond,
without any additional cofactors, can be envisaged (Fig. 13.15A). It is also possible
to propose potential mechanisms where
p-coumaryl coumarate (
54) [or other pcoumaryl
alcohol esters, e.g.,
p-coumaryl acetate (
52)] generates, through the
addition of an incoming hydride, chavicol (
31) and/or its regioisomer
p-anol
(
37) (Fig. 13.15B). Indeed, the second step in monolignol reduction was shown
to be the action of regiospecific reductases that transfer a hydride from NADH or NADPH into either the C-7 or the C-9 of the corresponding monolignol ester (or a
quinone methide derivative thereof), thus forming either an allyl or propenyl
phenol, respectively (Figs. 13.14 and 13.15B).
![FIGURE 13.15 Possible mechanisms for conversion of p-coumaryl alcohol esters into (A) hinokiresinol (56/57) and (B) chavicol (31) and p-anol (37). (A) (a) Concerted or (b) through intermediacy of a quinone methide and (B) (c) and (d) formation of a quinone methide intermediate through displacement of the (interchangeable) ester leaving group, with subsequent reduction by hydride [from NAD(P)H] and rearomatization to form either (c) chavicol (31) and/or (d) p-anol (37). The reactions in (B) may also proceed through direct displacement, without intermediacy of the quinone methide, by an incoming hydride at carbons 7 or 9 to form chavicol (31) or p-anol (37), respectively (not shown). Source: Modified from Vassão et al. (2006b).](images/f.13.15.jpg) |
| FIGURE 13.15 Possible mechanisms for
conversion of p-coumaryl alcohol esters
into (A) hinokiresinol
(56/57) and
(B) chavicol (31) and p-anol (37).
(A) (a) Concerted or (b) through
intermediacy of
a quinone methide and
(B) (c) and (d) formation of a quinone
methide intermediate through
displacement of the (interchangeable)
ester leaving group, with subsequent
reduction by hydride
[from NAD(P)H]
and rearomatization to form either (c)
chavicol (31) and/or (d) p-anol (37). The
reactions in (B) may also proceed
through direct displacement, without
intermediacy of the
quinone methide, by
an incoming hydride at carbons 7 or 9 to
form chavicol (31) or p-anol (37),
respectively (not shown). Source:
Modified from Vassão et al. (2006b). |
These regiospecific reductases (e.g., chavicol and eugenol synthase, CS/ES,
and isoeugenol synthase, IES) have been studied to a larger extent than the
monolignol-specific acyltransferases. Computational analyses of CS/ES isolated
from basil and IES isolated from petunia indicate greatest homology (~40–45%
identity) (Koeduka
et al., 2006) to members of the PIP family of reductases
(pinoresinol-lariciresinol, isoflavone, and phenylcoumaran-benzylic ether reductases)
we have either discovered and/or extensively characterized (Dinkova-
Kostova
et al., 1996; Fujita
et al., 1999; Gang
et al., 1999), and for which crystal
structures have been determined (Min
et al., 2003). Significantly, based on
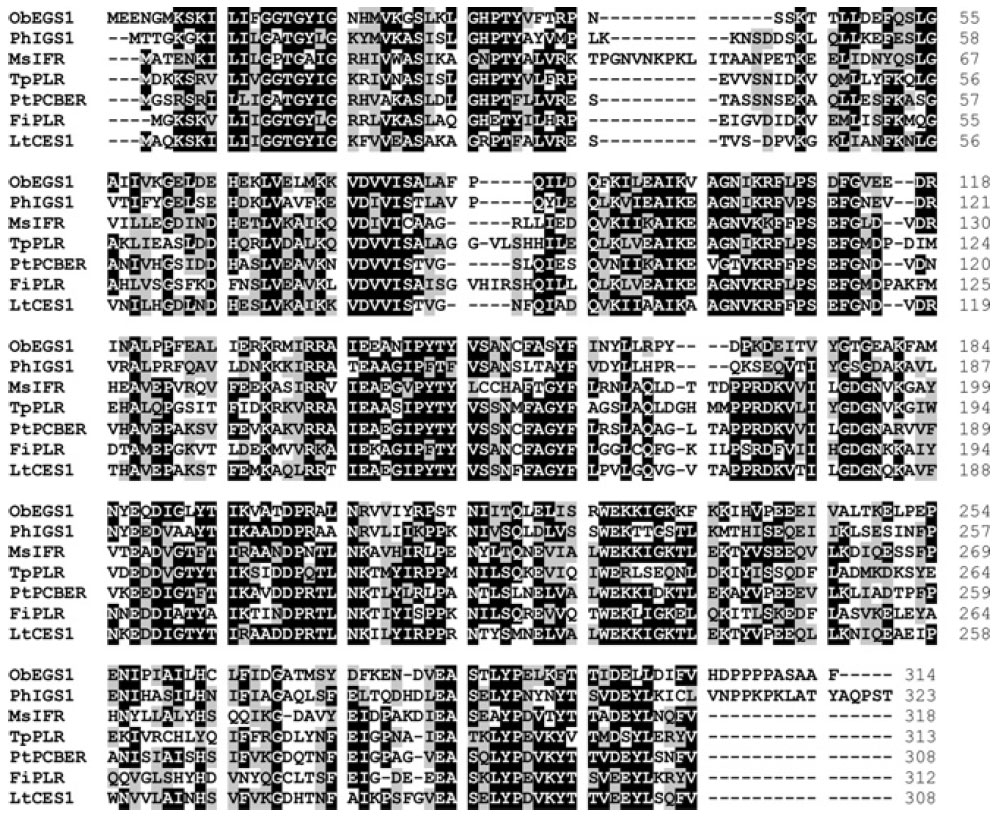
sequence homology, one such PLR/CS/ES homologue from
L. tridentata (LtCES1) was recently isolated and characterized (Fig. 13.16) (Vassão
et al., 2007).
While PLRs are involved in formation of other medicinally important plant
metabolites [e.g., podophyllotoxin (
45) and secoisolariciresinol (
46)] and various
plant defense compounds [e.g., plicatic acid (
30) in western red cedar heartwood],
the biochemical mechanisms of PLR, PCBER, and CS/ES share common properties,
including (1) a necessity for a free phenolic functionality in the substrate,
indicative of a common quinone methide intermediate (Figs. 13.14 and 13.17)
(Kim
et al., 2007; Koeduka
et al., 2006; Min
et al., 2003), and (
2) a highly conserved
Lys residue (K138 in PLR from
T. plicata, K133 in its homologue in
L. tridentata,
and K132 in CS/ES from basil) required for catalysis (Min
et al., 2003).
Figure 13.18
depicts the X-ray crystal structure of one member of this class of reductases, PLR
from
T. plicata (Min
et al., 2003). Based on the proposed catalytic mechanisms of
CS/ES (Koeduka
et al., 2006) and PLR (Min
et al., 2003), it is hardly surprising that
a high level of similarity was observed between these proteins. All of the PIP
reductases, as well as CS/ES, utilize NAD(P)H as the source of a hydride that is
regiospecifically (or stereospecifically) added to a carbon that originated from a
phenylpropanoid side chain (i.e., either a monolignol derivative or dimer). In fact,
a brief phylogenetic analysis indicates that these homologues cluster together
with PLR (e.g., from
T. plicata), PCBER (e.g., from
P. taeda), IFR (e.g., from
Medicago), and leucoanthocyanidin reductases (LACR, e.g., from
Vitis vinifera),
with the
L. tridentata homologue clustering closer to more distant PCBER and IFR
homologues (Fig. 13.19).
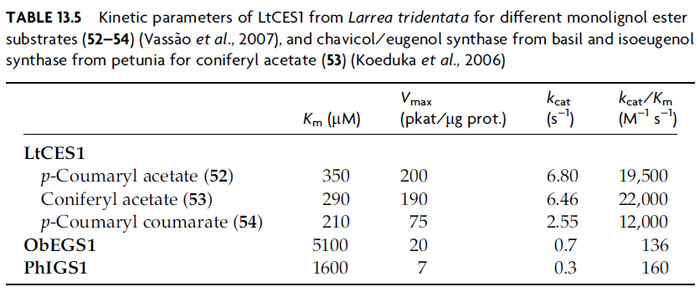
The biochemical characteristics of these enzymes have been studied, with basil
CS/ES and petunia IES reported to have substrate affinities [
Km, coniferyl acetate
(
53)] of 1.6–5.1 mM and
Vmax of 7–20 pkat/mg protein. These are indicative of
relatively low substrate affinity, although not far from the range of other enzymes
involved in volatile oil biosynthesis (Koeduka
et al., 2006). Additionally, the
corresponding PLR homologue in the creosote bush (
L. tridentata) catalyzes similar
conversions, but interestingly with apparently higher catalytic efficacy [
Km values of a few hundred micromolar and
Vmax values of a few hundred pkat/µg
protein for coniferyl acetate (
53),
p-coumaryl acetate (
52), and
p-coumaryl coumarate
(
54) (Vassão
et al., 2007); see Table 13.5]. We are currently examining the
properties of other PIP reductases regarding their abilities to form allyl and
propenyl phenols and have thus far seen evidence of some substrate versatility
in PLRs acting on monolignol esters (unpublished observations).
 |
| FIGURE 13.16 Amino acid alignment of basil
(Ocimum basilicum) chavicol/eugenol synthase
(ObEGS1), Petunia hybrida isoeugenol synthase
(PhIGS1), and PIP reductases from Medicago
sativa (MsIFR), Thuja plicata (TpPLR), Pinus
taeda (PtPCBER), Forsythia intermedia
(FiPLR),
and Larrea
tridentata (LtCES1). |
In effect, the long-standing question regarding the biochemical formation of
these widely used compounds, allyl and propenyl phenols, has now been elucidated
and shown to utilize the same pathway precursors as lignin biosynthesis.
Proteins (and their corresponding genes) involved in this process have been
isolated and characterized, thus presenting a new approach with which to study
and alter the lignification program of woody plants, as well as enabling the
production of these compounds in more commonly cultivated plants.



![FIGURE 13.15 Possible mechanisms for conversion of p-coumaryl alcohol esters into (A) hinokiresinol (56/57) and (B) chavicol (31) and p-anol (37). (A) (a) Concerted or (b) through intermediacy of a quinone methide and (B) (c) and (d) formation of a quinone methide intermediate through displacement of the (interchangeable) ester leaving group, with subsequent reduction by hydride [from NAD(P)H] and rearomatization to form either (c) chavicol (31) and/or (d) p-anol (37). The reactions in (B) may also proceed through direct displacement, without intermediacy of the quinone methide, by an incoming hydride at carbons 7 or 9 to form chavicol (31) or p-anol (37), respectively (not shown). Source: Modified from Vassão et al. (2006b).](images/f.13.15_large.jpg)