Biosynthesis of Monolignols
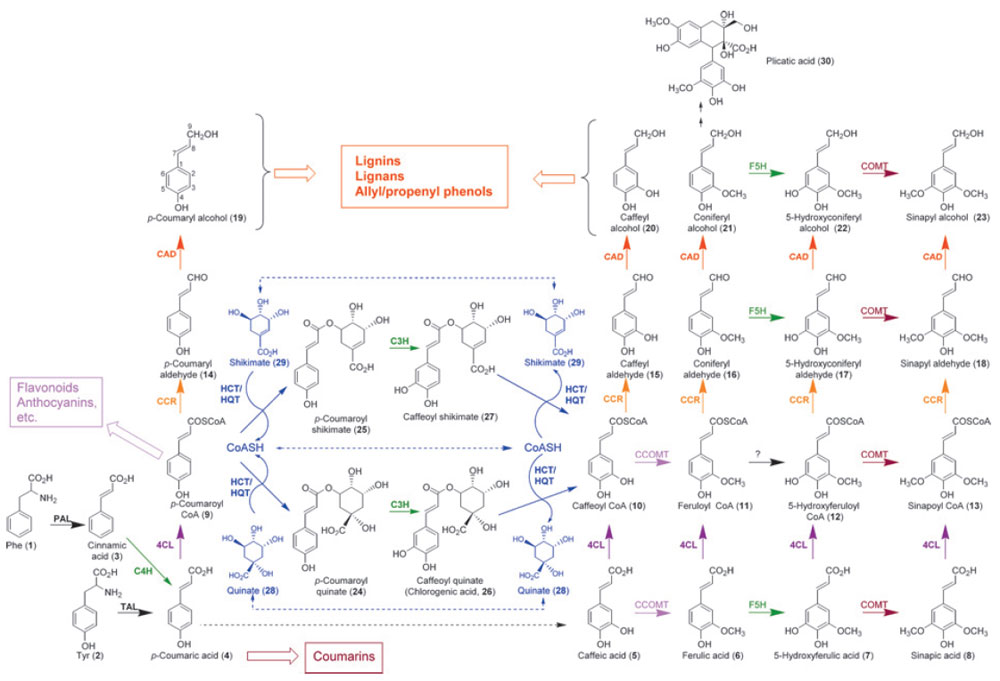
The phenylpropanoid pathway (Fig. 13.1) entry point is generally considered to be
the amino acid phenylalanine (
1, Phe), through action of phenylalanine ammonia
lyase (PAL, EC 4.3.1.5) forming
trans-cinnamic acid (
3). Some monocots (e.g.,
maize), though, are also able to utilize the
p-hydroxylated amino acid tyrosine
(
2, Tyr) as a partial entry point to this pathway through tyrosine ammonia lyase
(TAL). The mechanism of phenylalanine (
1) formation
in planta has been a matter
of debate, with arogenate decarboxylation/dehydration appearing to be the preferred
biosynthetic route (Cho
et al., 2007; Jung
et al., 1986). The subsequent
deamination reaction, catalyzed by PAL [forming cinnamic acid (
3) and stoichiometric
amounts of ammonium ion], was discovered by Eric Conn and his group
(Koukol and Conn, 1961) and is probably the most extensively studied reaction in
plant secondary metabolism (Lewis
et al., 1999).
The aromatic amino acid ammonia lyases (PAL, TAL, and HAL, histidine
ammonia lyase) all possess the interesting internal cofactor 3,5-dihydro-5-methylidene-4
H-imidazol-4-one (MIO) formed by spontaneous dehydration and cyclization
of a conserved tripeptide sequence (Ala-Ser-Gly) (Baedeker and Schulz,
2002; Calabrese
et al., 2004; Schwede
et al., 1999), which acts as a strong electrophile
during catalysis. The mechanism of ammonia elimination is still controversial,
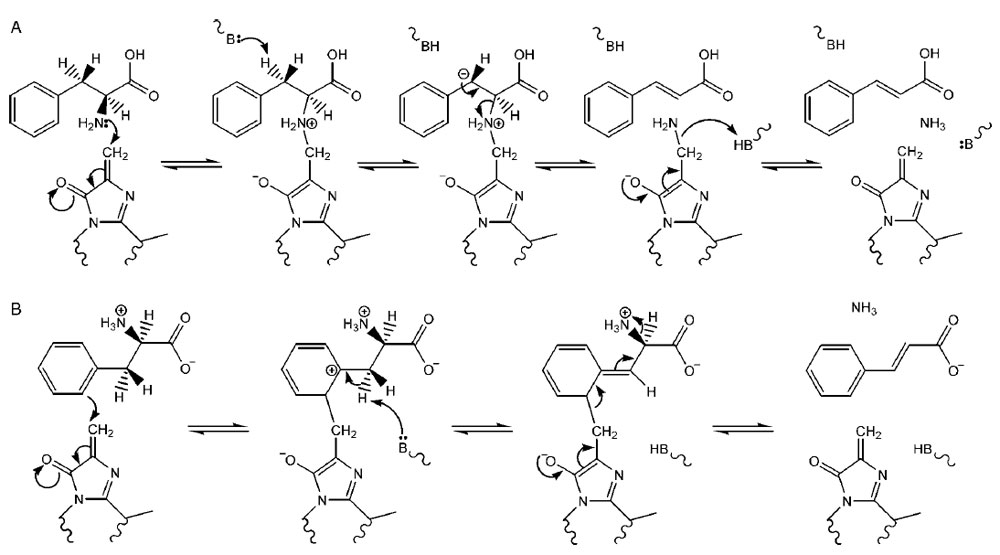
with the two most likely possibilities currently being (
1) a E1cb-like
mechanism where MIO forms a bond to the amine group of the amino acid
(Fig. 13.3A) or (
2) a Friedel-Crafts-like mechanism where MIO reacts with the
aromatic ring of the substrate, which therefore loses aromaticity and increases the
acidity of the nearby β-proton (Fig. 13.3B) (Calabrese
et al., 2004; Louie
et al., 2006; Poppe and Rétey, 2005). Recent studies of TAL from
Rhodobacter sphaeroides have
determined
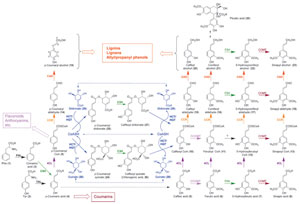 |
| FIGURE 13.1 Current view of the
phenylpropanoid pathway. 4CL,
hydroxycinnamoyl CoA ligases; C3H,
p-coumarate 3-hydroxylase; C4H, cinnamate
4-hydroxylase; CAD, cinnamyl alcohol
dehydrogenases; CCOMT, hydroxycinnamoyl
CoA O-methyltransferases; CCR, cinnamoyl
CoA oxidoreductases;
COMT, caffeic acid
O-methyltransferase; F5H, ferulate
5-hydroxylase; HCT/HQT, hydroxycinnamoyl
shikimate/quinate transferase; PAL,
phenylalanine
ammonia lyase; TAL, tyrosine ammonia lyase. |
that Tyr (
2) preference (i.e., TAL activity) is largely dictated by one
specific TAL-conserved active site residue, H89, which is thought to be hydrogenbonded
to the phenolic moiety of the substrate Tyr (
2). When H89 is replaced with
a Phe residue (characteristic of PAL), substrate selectivity is switched and the
mutant H89F TAL
prefers Phe (1) (i.e., PAL activity). When the corresponding
inverse mutation was made in PAL from
Arabidopsis thaliana, the mutant (F144H)
PAL enzyme lost PAL activity and instead gained TAL activity (Louie
et al., 2006;
Watts
et al., 2006).
Although initially regarded as a rate-determining reaction in the pathway
(Camm and Towers, 1973; Rubery and Fosket, 1969), PAL does not seem to
serve as a major carbon allocation regulator, with flux apparently being determined
by the availability of Phe (
1) and by downstream enzymes in the pathway,
especially C4H/C3H (Anterola
et al., 1999, 2002). In
A. thaliana, a four-membered
PAL multigene family (
AtPAL1–4) has been characterized, with Tyr (
2) serving, as
expected, as a very poor substrate. Kinetic parameters for AtPAL1, 2, and 4 with
Phe (
1) were similar, with
Km values of ~60–70 µM and
V
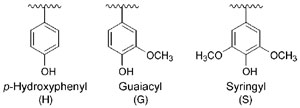 |
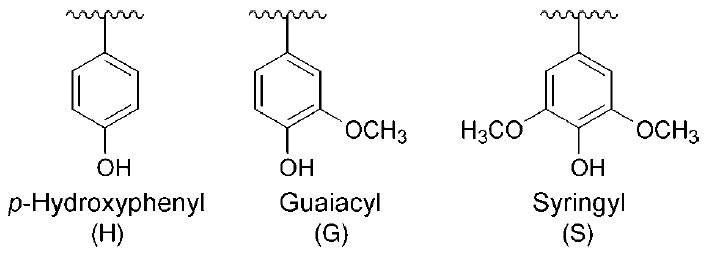
| FIGURE 13.2 Aromatic (H, G, and S) residues
in plant lignins. |
max ~5–10 pkat/µg
protein, whereas AtPAL3 was barely active (
Km = 2.56 mM,
Vmax = 0.4 pkat/µg
protein) (Cochrane
et al., 2004). Rohde
et al. (2004) have also analyzed the effects of
PAL downregulation in
A. thaliana. In particular,
pal1 and
pal2 mutants apparently
presented no clear phenotypic alterations, with lignin levels of approximately
60–70% of wild type (WT), perhaps as a result of compensatory upregulation of
the remaining PAL genes in each mutant (i.e.,
AtPAL2/4 were upregulated in pal1
mutants, AtPAL1/4 in pal2 mutants). Double
pal1
pal2 mutants, on the other hand,
exhibited approximately 25% of WT PAL activity (resulting mainly from the
presence of AtPAL4), were sterile, and had estimated lignin levels of approximately
30–35% of WT, with this lignin having its S/G ratio apparently nearly
doubled relative to WT.
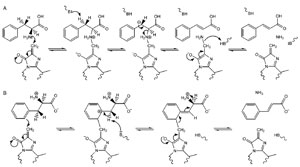 |
| FIGURE 13.3 Potential mechanisms for PAL
catalysis: (A) E1cb-like mechanism and
(B) Friedel-Crafts-like mechanism. Source:
Redrawn from
Calabrese et al. (2004). |
Trans-cinnamic acid (
3) is next para-hydroxylated by cinnamate 4-hydroxylase
(C4H, EC 1.14.13.11), a membrane-associated cytochrome
p-450 hydroxylase
acting along with an associated NADPH-dependent reductase (Lewis
et al.,
1999), to afford
p-coumaric acid (
4). Subsequent ring hydroxylation reactions
[i.e., C3H-catalyzed 3-hydroxylation leading to formation of the catechol-like
substructure of caffeic acid (
5) and derivatives, as well as F5H-catalyzed
5-hydroxylation leading to the 5-hydroxyguaiacyl substructure present in
5-hydroxyconiferyl alcohol (
22)] are also performed by cytochrome
p-450 hydroxylases.
Therefore, these enzymes (C4H, C3H, and F5H) are often grouped
together as a single class. In
A. thaliana, each of these enzymes is encoded by either
one- (C4H and C3H) or two-membered (F5H) gene families, with locus numbers
At2g30490 (C4H), At2g40890 (C3H), and At4g36220/At5g04330 (F5H), respectively.
As of yet, no X-ray crystal structures for any of these plant cytochrome
p-450 enzymes have been reported, although they play important (i.e., possibly
regulatory, in the case of C4H and C3H) roles in monolignol biosynthesis
and lignification.
Interestingly, 3-hydroxylation of the
p-coumaric acid (
4) moiety apparently
occurs only after esterification, first to its CoA form (see below), then to shikimate and/or quinate esters; that is,
p-coumarate 3-hydroxylase (C3H) acts on
p-coumaroyl quinate (
24) and/or shikimate (
25) to form the corresponding caffeic
acid derivative (26 and/or 27, respectively). Recognition that the
p-coumaroyl
esters serve as substrates for cytochrome
p-450 hydroxylation was established
using parsley (Petroselinum crispum) cell suspension cultures (Heller and Kühnl,
1985); more recent studies identified the gene and expressed functional recombinant
protein (Schoch
et al., 2001). In this regard, transesterases (HCT/HQT) that
transfer hydroxycinnamoyl groups from the corresponding CoA esters to shikimate
(
29) and quinate (
28), respectively, were also recently reported (Hoffmann
et al., 2003; Niggeweg
et al., 2004). HCT, which prefers shikimate (
29) as an acyl
acceptor, has been reported to be present in stem vascular tissues in tobacco,
silencing of which in
A. thaliana led to dwarfed plants. In HCT-silenced Nicotiana
benthamiana plants, the corresponding lignin reportedly decreased and presented
lower S and increased H contents, respectively, that is, in agreement with its
presumed participation in monolignol biosynthesis (Hoffmann
et al., 2004).
The final ring hydroxylation catalyzed by the cytochrome
p-450 F5H was first
reported to act on ferulic acid (
6) (Grand, 1984); however, findings by Fukushima
and colleagues indicated that the hydroxylation step preferentially involved coniferyl
alcohol (
21) (Chen
et al., 1999a,b). The corresponding F5H (
CYP84) gene was
subsequently isolated from
A. thaliana (Meyer
et al., 1996), and further analysis of
the recombinant CYP84 protein indicated a preference for both coniferyl alcohol
(
21) and coniferyl aldehyde (
16) rather than ferulic acid (
6) as substrates [
Km values
of 3 and 1 µM,
Vmax of 6 and 5 pkat/mg protein for 21 and 16, respectively, while
Km = 1 mM and
Vmax = 4 pkat/mg protein for the latter (
6)] (Humphreys
et al.,
1999), which is in agreement with Fukushima’s seminal findings.
Through further modification of the aromatic ring, the catechol-like substructure
of caffeic acid (
5) [or an ester derivative thereof, e.g., caffeoyl CoA (
10)],
caffeyl aldehyde (
15), and/or caffeyl alcohol (
20) can serve as substrate for SAM
(S-adenosyl methionine)-dependent
O-methyltransferases (OMTs). In this way,
the guaiacol substructure of ferulic acid (
6) [or the respective ester derivative, e.g.,
feruloyl CoA (
11)], coniferyl aldehyde (
16), and/or coniferyl alcohol (
21), respectively,
can be formed. This
O-methylation was originally reported to be performed
by the so-called caffeic acid O-methyltransferases (COMTs) but was later shown
to much more likely utilize caffeoyl CoA (
10) as substrate i.e., caffeoyl CoA
O–methyltransferase (CCOMT) (Pakusch
et al., 1989; Schmitt
et al., 1991; Ye
et al., 1994). Analogously, the 5-hydroxyguaiacol substructure resulting from
F5H-mediated hydroxylation is
O-methylated to afford a syringyl moiety. This
is catalyzed by an OMT originally thought to act on caffeic acid (
5) as primary
substrate (COMT), and although the enzyme has now been established to be a
5-hydroxyguaiacyl
O-methyltransferase (Anterola and Lewis, 2002; Atanassova
et al., 1995), the original ‘‘COMT’’ denomination still largely remains in use.
In
A. thaliana, 22 genes have been putatively annotated as either COMT
(EC 2.1.1.68, 17 putative genes) or CCOMT (EC 2.1.1.104, 5 putative genes).
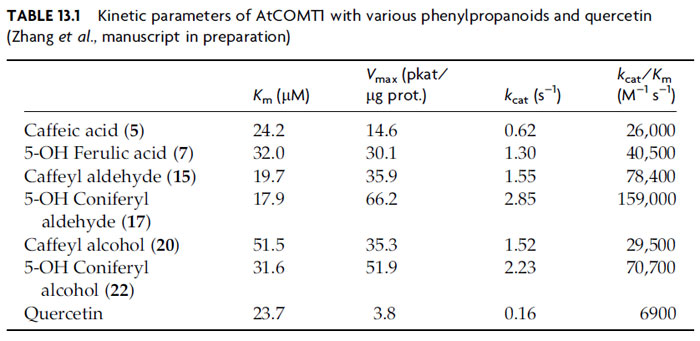
Biochemical analysis of 12 putative AtCOMT proteins bearing highest homology
to the previously characterized AtCOMT1 indicated that only the latter is able to
efficiently
O-methylate a variety of potential substrates, with the remaining 11 not able to act on any of the phenolic compounds tested (Zhang
et al., manuscript in
preparation). Caffeyl (
15) and 5-hydroxyconiferyl (
17) aldehydes are the preferred
substrates, having
kcat/K
m values of 78,400 and 159,000 M
-1 s
-1, respectively
(Table 13.1). This is in accordance with previous analyses (see Anterola and
Lewis, 2002), where the largest effect of COMT downregulation was the decrease
of S contents in lignin (whereas G levels and total lignin amounts apparently
remained relatively unchanged), with the concomitant incorporation of
5-hydroxyconiferyl alcohol (
22) into the mutated lignin resulting in weakened
and discolored stems. Interestingly, AtCOMT1 has also been described as an
enzyme involved in quercetin to isorhamnetin formation (Muzac
et al., 2000);
however, the kinetic data indicate that quercetin is a poorer substrate by an
order of magnitude (Table 13.1) (Zhang
et al., manuscript in preparation).
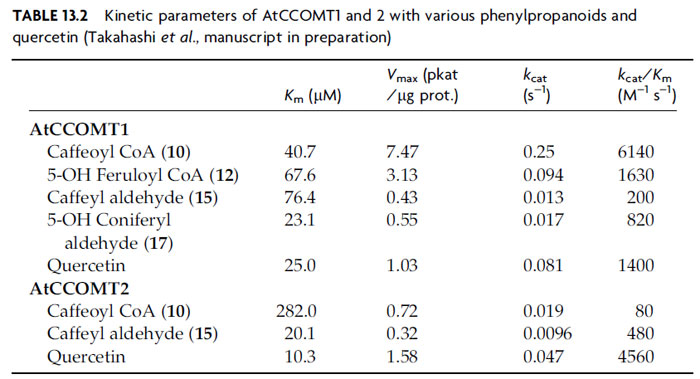
Correspondingly, work on the five putative AtCCOMT enzymes has thus
far focused on AtCCOMT1 and 2. AtCCOMT1 uses caffeoyl CoA (
10) and
5-hydroxyferuloyl CoA (
12) as preferred substrates (Table 13.2), together with
quercetin, although with efficacies 10–100-fold lower than those observed for
AtCOMT1 acting on the corresponding substrates [caffeyl aldehyde (
15) and
5-hydroxyconiferyl aldehyde (
17), respectively]. The corresponding acids, as
expected, did not serve efficiently as substrates. AtCCOMT2, by contrast,
more efficiently methylated quercetin, rather than the possible monolignol
pathway intermediates, thus suggesting a role in flavonoid metabolism. Interestingly,
AtCCOMT2-catalyzed methylation occurred at both positions 4 and 5
with 5-hydroxyferuloyl CoA (
12) and 5-hydroxyconiferyl aldehyde (
17)
(Takahashi
et al., manuscript in preparation), indicating that it has no role in
monolignol formation, where position 4 remains unmodified. In summary, while
the participation of AtCOMT1 in monolignol biosynthesis in
A. thaliana appears to
be quite well established (i.e., S monomer formation), the particular roles of the
multiple AtCCOMT isoforms, and the particular substrates they act upon, are still
a matter of debate and thus remain to be further determined.
The transformations occurring on the phenolic ring (e.g., hydroxylations,
O-methylations) are accompanied by side-chain modification of the aforementioned cinnamic acid derivatives (
4–8), first generating CoA esters (
9–13) through the twostep
activity of hydroxycinnamoyl CoA ligases (4CL, EC 6.2.1.12). These utilize ATP
to form an AM
p-acyl intermediate that stays enzyme-bound, with AMP then enzymatically
displaced by CoA. In
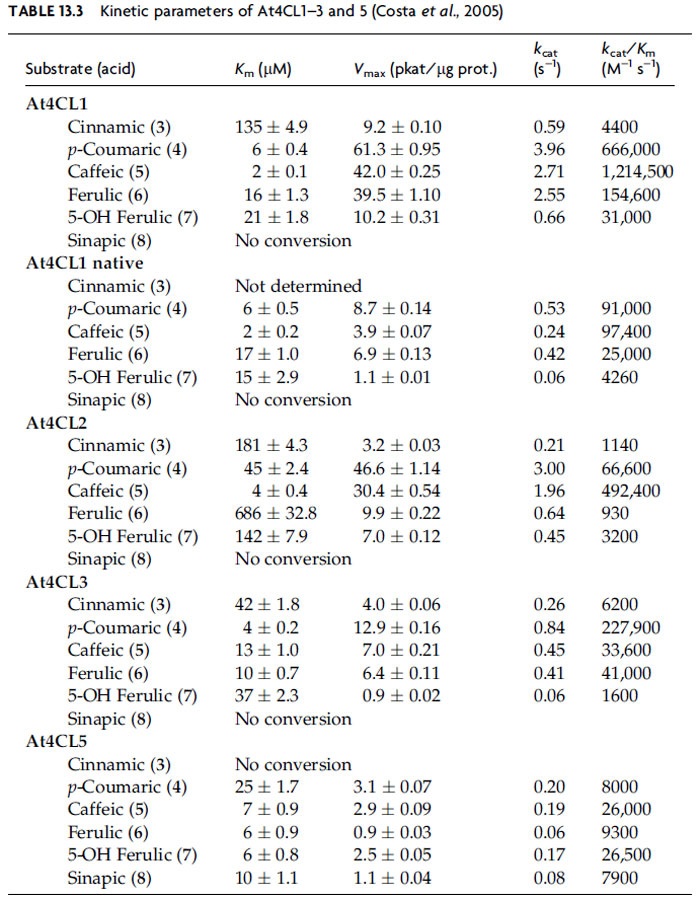
A. thaliana, 14 genes had been originally annotated as
putative 4CLs, but through further analysis and biochemical characterization, only 4
genes were shown to encode functional 4CL enzymes, albeit with very distinct
kinetic properties [using all six possible substrates, cinnamic (3),
p-coumaric (4),
caffeic (5), ferulic (6), 5-hydroxyferulic (7), and sinapic (8) acids]. At4CL1 is by far the
most active, with
kcat/
Km being ~15–600 times higher than that for others
(Table 13.3) (Costa
et al., 2005).
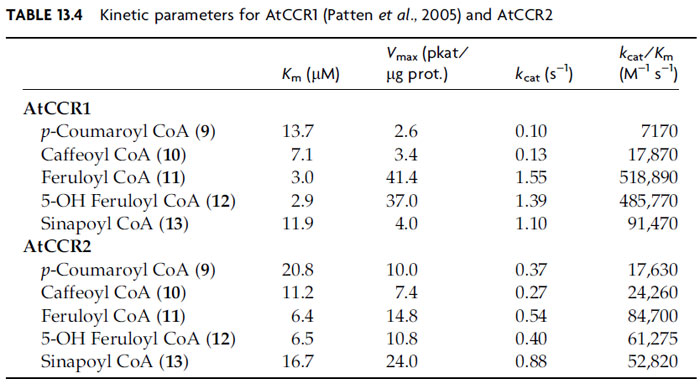
The hydroxycinnamoyl CoA esters (
9–13) thus formed serve as substrates for
reduction by NADPH-dependent cinnamoyl CoA oxidoreductases (CCR, EC
1.2.1.44) generating the corresponding aldehydes (
14–18), with 11 genes being
putatively annotated as CCRs in
A. thaliana. To date, only three of the putative
AtCCRs have been biochemically characterized, namely, AtCCR1, AtCCR2
(Lauvergeat
et al., 2001; Patten
et al., 2005), and AtCCR8. The most active of these
is AtCCR1, which most effectively utilized feruloyl CoA (
11) and
5-hydroxyferuloyl CoA (
12) as substrates (Table 13.4) (Patten
et al., 2005).
This is in harmony with the corresponding aldehydes [coniferyl (
16) and
5-hydroxyconiferyl (
17) aldehydes] serving as substrates for subsequent monolignol
formation. AtCCR2, by contrast, was a less effective catalyst, but again
slightly preferred compounds 11/12 as substrates (Table 13.4). Interestingly,
AtCCR8 displays both CCR and CAD catalytic properties, being able to slowly
convert cinnamyl,
p-coumaryl (
14), caffeyl (
15), and sinapyl (
18) aldehydes into the
corresponding monolignols; however, the physiological significance of these latter
observations is still unclear, given the substrate versatility of many proteins.
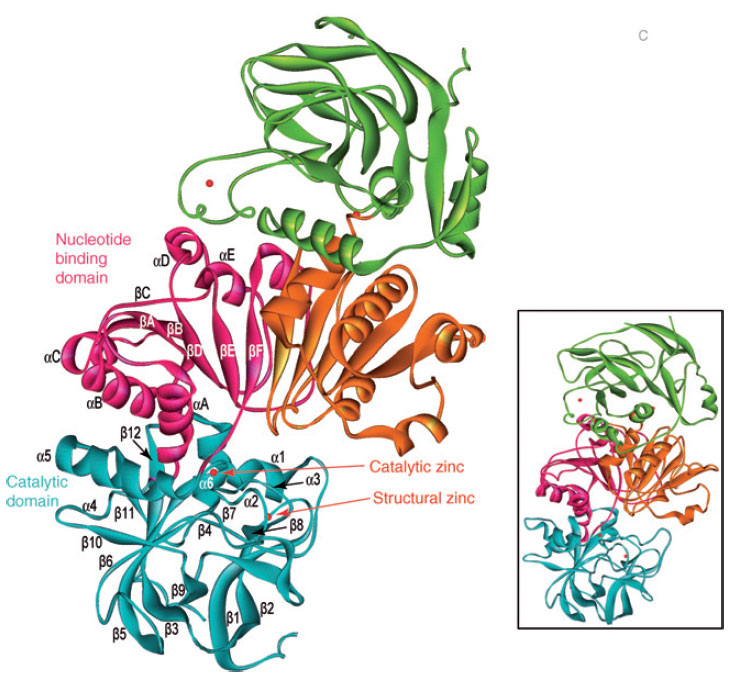
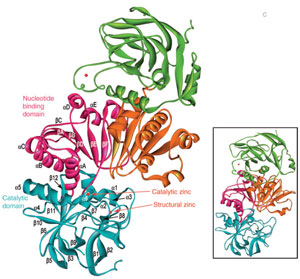 |
| FIGURE 13.4 Schematic representation of the
crystal structure of the AtCAD5 homodimer and
energy-minimized model of AtCAD4 (inset). |
The hydroxycinnamyl aldehydes (14–18) are then further reduced (by cinnamyl
alcohol dehydrogenases, CAD, EC 1.1.1.195) to afford the monolignols 19–23 (Lewis
et al., 1999). These NADPH-dependent CADs thus catalyze the final step in
monolignol biosynthesis and have been studied in some detail in
A. thaliana and
other plants. In
A. thaliana,
in silico analyses had indicated the existence of a
17-membered CAD family, but further biochemical examination established that
2 of those (AtCAD4/5) were catalytically the most active (Kim
et al., 2004). Moreover, none of the isoforms displayed any strong sinapyl aldehyde (
18)
preference, contrary to reports of a putative sinapyl alcohol dehydrogenase
(SAD) specific for sinapyl aldehyde (
18) (Li
et al., 2001). X-ray crystal structures
for AtCAD4 and 5 have been recently determined (Fig. 13.4), with each being a
dimer containing two zinc ions per subunit, one participating in catalysis and the
other playing a structural role (Youn
et al., 2006a). An
A. thaliana cad4
cad5 double
mutant has also been obtained (Jourdes
et al., 2007; Sibout
et al., 2005) and presents
a visible prostrate phenotype (i.e., cannot stand upright, see Fig. 13.5A) after
approximately 4 weeks growth, resulting from the large decrease in amounts of
lignin proper. Furthermore, this double mutant produces only about approximately
10% of WT lignin amounts, based on monolignol-derived
thioacidolysis
cleavage data, with small amounts of a poly-
p-hydroxycinnamyl aldehyde also
being formed. Interestingly, deposition of the latter is prematurely aborted relative
to lignin proper (Jourdes
et al., 2007). This further emphasizes the stringency
of the lignification process, where three of the CAD monolignol products (19, 21,
and 23) have evolutionarily become the highly conserved substrates for
subsequent controlled one electron (radical) oxidation and polymerization of the
H, G, and S ‘‘building blocks’’ of lignin (Fig. 13.2) (Anterola and Lewis, 2002). That
is, strong evolutionary pressure resulted in lignins being formed from monolignols
(as well as, to a much lesser extent, monolignol esters in grasses).