Quinoline Alkaloids
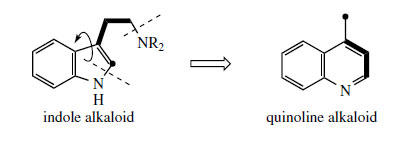
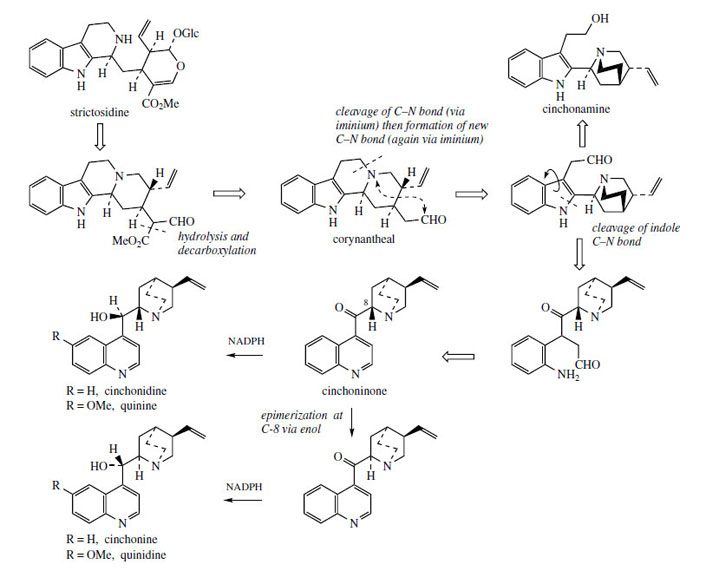
Some of the most remarkable examples of terpenoid indole alkaloid modifications are to be found in the genus Cinchona* (Rubiaceae), in the alkaloids quinine, quinidine, cinchonidine, and cinchonine (Figure 88), long prized for their antimalarial properties. These structures are remarkable in that the indole nucleus is no longer present, having been rearranged into a quinoline system (Figure 89). The relationship was suspected quite early on, however, since the indole derivative cinchonamine (Figure 90) was known to co-occur with these quinoline alkaloids. An outline of the pathway from the Corynanthe-type indole alkaloids to cinchonidine is shown in Figure 90. The conversion is dependent on the reversible processes by which amines plus aldehydes or ketones, imines (Schiff bases), and their reduction products are related in nature. Suitable modification of strictosidine leads to an aldehyde (compare the early reactions in the ajmalicine pathway (Figure 76)).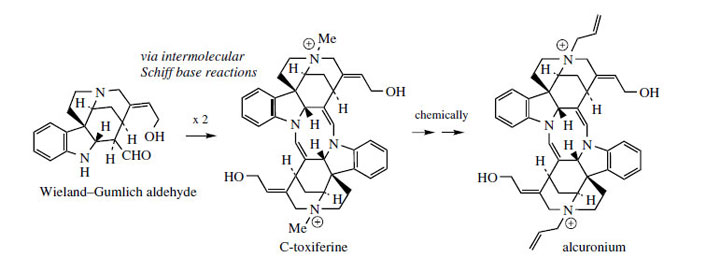 |
| Figure 85 |
 |
| Figure 86 |
Ellipticine
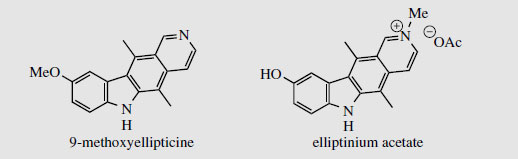
Ellipticine (Figure 86) and related alkaloids, e.g. 9-methoxyellipticine (Figure 87), are found in the bark of Ochrosia elliptica (Apocynaceae) and other Ochrosia species. Clinical trials with these alkaloids and a number of synthetic analogues showed them to be potent inhibitors of several cancerous disorders, but pre-clinical toxicology indicated a number of side-effects, including haemolysis and cardiovascular effects. Ellipticines are planar molecules that intercalate between the base pairs of DNA and cause a partial unwinding of the helical array. There is a some correlation between the degree of unwinding and the biological properties, those showing the largest unwinding inhibiting the greatest number of cancerous cells. Recent research suggests there may be more than one mechanism of action, however. Ellipticine is oxidized in vivo mainly to 9-hydroxyellipticine, which has an increased activity, and it is believed that this may in fact be the active agent. Poor water-solubility of ellipticine and derivatives gave problems in formulation for clinical use, but quaternization of 9-hydroxyellipticine to give the water-soluble 9-hydroxy-2-N-methylellipticinium acetate (elliptinium acetate) (Figure 87) has produced a highly active material, of value in some forms of breast cancer, and perhaps also in renal cell cancer. A variety of such quaternized derivatives is being tested, and some water-soluble N-glycosides also show high activity.
 |
| Figure 87 |
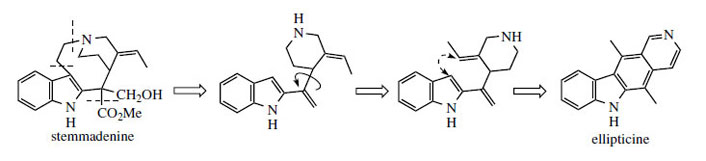
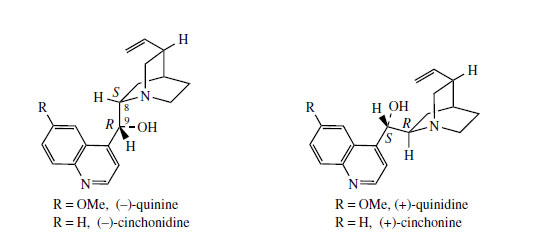
Hydrolysis/decarboxylation would initially remove one carbon from the iridoid portion and produce corynantheal.An intermediate of the cinchonamine type would then result if the tryptamine side-chain were cleaved adjacent to the nitrogen, and if this nitrogenwere then bonded to the acetaldehyde function. Ring opening in the indole heterocyclic ring could generate new amine and keto functions. The new heterocycle would then be formed by combining this amine with the aldehyde produced in the tryptamine side-chain cleavage. Finally, reduction of the ketone gives cinchonidine or cinchonine. Hydroxylation and methylation at some stage allows biosynthesis of quinine and quinidine. Quinine and quinidine, or cinchonidine and cinchonine, are pairs of diastereoisomers, which have opposite chiralities at two centres (Figure 88). Stereospecific reduction of the carbonyl in cinchoninone can control the stereochemistry adjacent to the quinoline ring (C-9). The stereochemistry at the second centre (C-8) is also determined during the reduction step, presumably via the enol form of cinchoninone (Figure 90).
 |
| Figure 88 |
 |
| Figure 89 |
 |
| Figure 90 |
Cinchona
Cinchona bark is the dried bark from the stem and root of species of Cinchona (Rubiaceae), which are large trees indigenous to South America. Trees are cultivated in many parts of the world, including Bolivia, Guatemala, India, Indonesia, Zaire, Tanzania, and Kenya. About a dozen different Cinchona species have been used as commercial sources, but the great variation in alkaloid content, and the range of alkaloids present, has favoured cultivation of three main species, together with varieties, hybrids, and grafts. Cinchona succirubra provides what is called 'red' bark (alkaloid content 5-7%), C. ledgeriana gives 'brown' bark (alkaloid content 5-14%), and C. calisaya 'yellow' bark with an alkaloid content of 4-7%. Selected hybrids can yield up to 17% total alkaloids. Bark is stripped from trees which are about 8-12 years old, the trees being totally uprooted by tractor for the process.
A considerable number of alkaloids have been characterized in cinchona bark, four of which account for some 30-60% of the alkaloid content. These are quinine, quinidine, cinchonidine, and cinchonine, quinoline-containing structures representing two pairs of diastereoisomers (Figure 88). Quinine and quinidine have opposite configurations at two centres. Cinchonidine and cinchonine are demethoxy analogues, but unfortunately use of the -id- syllable in the nomenclature does not reflect a particular stereochemistry. Quinine is usually the major component (half to two-thirds total alkaloid content) but the proportions of the four alkaloids vary according to species or hybrid. The alkaloids are often present in the bark in salt combination with quinic acid or a tannin material called cinchotannic acid. Cinchotannic acid decomposes due to enzymic oxidation during processing of the bark to yield a red pigment, which is particularly prominent in the 'red' bark.
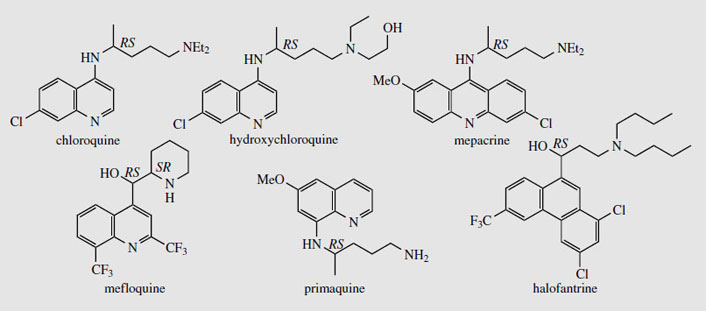
Cinchona and its alkaloids, particularly quinine, have been used for many years in the treatment of malaria, a disease caused by protozoa, of which the most troublesome is Plasmodium falciparum. The beneficial effects of cinchona bark were first discovered in South America in the 1630s, and the bark was then brought to Europe by Jesuit missionaries. Religious intolerance initially restricted its universal acceptance, despite the widespread occurrence of malaria in Europe and elsewhere. The name cinchona is a mis-spelling derived from Chinchon. In an often quoted tale, now historically disproved, the Spanish Countess of Chinchon, wife of the viceroy of Peru, was reputedly cured of malaria by the bark. For many years, the bark was obtained from South America, but cultivation was eventually established by the English in India, and by the Dutch in Java, until just before the Second World War, when almost all the world's supply came from Java. When this source was cut off by Japan in the Second World War, a range of synthetic antimalarial drugs was hastily produced as an alternative to quinine. Many of these compounds were based on the quinine structure. Of the wide range of compounds produced, chloroquine, primaquine, and mefloquine (Figure 91) are important antimalarials. Primaquine is exceptional in having an 8-aminoquinoline structure, whereas chloroquine and mefloquine retain the 4- substituted quinoline as in quinine. The acridine derivative mepacrine (Figure 91), though not now used for malaria treatment, is of value in other protozoal infections. Halofantrine(Figure 91) dispenses with the heterocyclic ring system completely, and is based on phenanthrene. At one time, synthetic antimalarials had almost entirely superseded natural quinine, but the emergence of Plasmodium falciparum strains resistant to the synthetic drugs, especially the widely used prophylactic chloroquine, has resulted in reintroduction of quinine. Mefloquine is currently active against chloroquine-resistant strains, but, whilst ten times as active as quinine, does produce gastrointestinal upsets and dizziness, and can trigger psychological problems such as depression, panic, or psychosis in some patients. The ability of P. falciparum to develop resistance to modern drugs means malaria still remains a huge health problem, and is probably the major single cause of deaths in the modern world. Chloroquine and its derivative hydroxychloroquine (Figure 91), although antimalarials, are also used to suppress the disease process in rheumatoid arthritis.
 |
| Figure 91 |
Quinine (Figure 88), administered as free base or salts, continues to be used for treatment of multidrug-resistantmalaria, though it is not suitable for prophylaxis. The specific mechanism of action is not thoroughly understood, though it is believed to prevent polymerization of toxic haemoglobin breakdown products formed by the parasite. Vastly larger amounts of the alkaloid are consumed in beverages, including vermouth and tonic water. It is amusing to realize that gin was originally added to quinine to make the bitter antimalarial more palatable. Typically, the quinine dosage was up to 600 mg three times a day. Quinine in tonic water is now the mixer added to gin, though the amounts of quinine used (about 80 mg l−1) are well below that providing antimalarial protection. Quinine also has a skeletal muscle relaxant effect with a mild curare-like action. It thus finds use in the prevention and treatment of nocturnal leg cramp, a painful condition affectingmany individuals, especially the elderly.
 |
| Figure 92 |
 |
| Figure 93 |
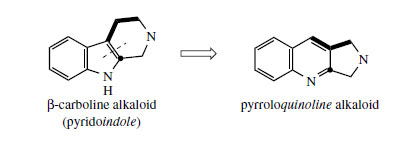
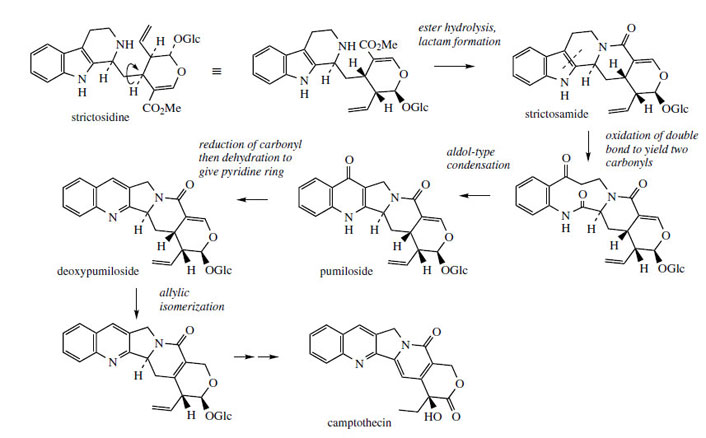
Camptothecin* (Figure 93) from Camptotheca acuminata (Nyssaceae) is a further example of a quinoline-containing structure that is actually derived by modification of an indole system. The main rearrangement process is that the original β- carboline 6–5–6 ring system becomes a 6–6–5 pyrroloquinoline by ring expansion of the indole heterocycle (Figure 92). In camptothecin, the iridoid portion from strictosidine is effectively still intact, the original ester function being utilized in forming an amide linkage to the secondary amine. This occurs relatively early, in that strictosamide is an intermediate. Pumiloside(also isolated from C. acuminata) and deoxypumilosideare potential intermediates. Steps beyond are not yet defined, but involve relatively straightforward oxidation and reduction processes (Figure 93).
Support our developers




