Immuno isolation of Organelles Using Magnetic Solid Supports
To study the molecular composition and functional properties of cellular organelles, these must first be purified. Classical subcellular fractionation methods are based on the physical properties of membrane-bound compartments, e.g., their density. However, many compartments have similar properties and cannot be separated from each other by such methods. Immunoisolation, however, allows you to affinity purify organelles using an antibody directed against a component of the compartment (Jones et al., 1998; Howell et al., 1989).
To set up an immunoisolation protocol, you first have to identify an appropriate antigen that is tightly associated with the membrane (or, even better, transmembrane) and localizes exclusively to the compartment that you wish to isolate. If the distribution of the antigen is broader, you might be able to remove unwanted organelles that contain this antigen by a prepurification step. Moreover, the antigen should be relatively abundant and its antigenic site should be well exposed on the cytoplasmic surface of the compartment.
Next, you need a high-affinity antibody that recognizes the native form of the chosen antigen. Because large amounts of the antibody may be needed to complete a set of experiments, it is better to use a monoclonal antibody whenever possible.
Finally, you have to choose a solid support. Of all the solid supports that we have tested, magnetic beads are the most convenient because of two key features: their smooth surface with low hydrophobidity ensures low nonspecific binding and they can be retrieved with a magnet, which minimizes membrane shearing when resuspending the beads.
Immunoisolated fractions can be analyzed by virtually any means, as the beads themselves are inert. A small hinderance is caused by the high abundance of antibodies in the samples, which requires the use of metabolically labeled fractions to establish the general protein pattern. Most types of analysis, including Western blotting, bideminsional gel electrophoresis, lipid analysis, electron microscopy, or enzymatic assays, can be performed using standard protocols. The main limitation is that only small amounts of membranes can be isolated, so it is, for example, not possible to microsequence a protein directly from an immunoisolated fraction.
II. MATERIALS AND INSTRUMENTATION
Cell scrapers (flexible rubber policemen) with a silicon rubber piece of about 2 cm, cut at a sharp angle, and attached to a metal bar can be made by your institute workshop. You need a standard low-speed cell centrifuge, Eppendorf centrifuge, and Beckman ultracentrifuges and rotors. Magnets can be purchased from Dynal Biotech (Cat. No. 120.20). A rotating wheel (e.g., Cat. No. 34526; Snijders) with a speed of about 2 rotations per minute should be used.
M450 sheep anti-mouse IgG Dynabeads (Cat. No. 110.01) and M450 goat anti-mouse IgG Dynabeads (Cat. No. 110.05) are manufactured by Dynal Biotech. It might be useful to buy Dynabeads in larger batches, as variation could occur between different lots. We find that M500 beads, which have been developed specifically for subcellular fractionation, work less well for our purposes than M450 beads, which are designed for cell isolation.
For the immunoisolation protocol described here, MDCK II cells were stably transfected with the pCB6 plasmid containing the human TfR with (mhTfR cells) or without (hTfR cells) a single myc tag (13 amino acids) at its cytoplasmic amino terminus. Transfection was carried out by the calcium phosphate method and was followed by selection with geneticin-sulphate (Invitrogen; Cat. No. 11811). Transfected cells should not be used for more than 8-10 passages to avoid loss of expression.
III. PROCEDURES
A. Postnuclear Supernatant
Any subcellular fractionation protocol starts with breaking the plasma membrane to release intact intracellular organelles (Howell et al., 1989). This can be achieved through several methods, which are all compatible with immunoisolation. For MDCK cells, we use passage through a needle in a slightly hypotonic medium.
- Briefly rinse the cells with ice-cold phosphatebuffered saline (PBS). Add 2.5 ml of ice-cold PBS to the dish. If MDCK cells grown on polycarbonate filters are used, add 2.5 ml of PBS to the apical side of the cells and transfer the filter to the inner side of the cool lid of the original filter support. Scrape the cells with a rubber policeman, taking care not to break them.
- Transfer to a 15-ml Falcon tube and centrifuge at 900 rpm for 5 min.
- Resuspend gently in 2.5ml of HB (250mM sucrose, 3 mM immidazole, pH 7.4) per tube and centrifuge at 2500 rpm for 10 rain.
- Add 150µl of HB+ (HB, 1 mM EDTA, 1µg/ml leupeptine, 1 µg/ml pepstatin, 1 µg/ml aprotinin, 100µg/ml phenymethylsulfonyl fluoride) per filter and resuspend gently with a blue tip approximately eight times. Do not apply the tip to the bottom of the tube, but on the side, to minimize cell breakage while resuspending the cells. Homogenize with a 1-ml syringe and a 22-gauge needle one to three times, depending on the state of the nuclei as observed under a microscope. Nuclei should be clean of cellular material and intact (round and dark grey).
- Dilute with 100µl of HB+ per filter to facilitate pelleting of the nuclei and centrifuge at 2500rpm for 10 min.
- Preclear the postnuclear supernatant (PNS) 5 min at 13,000rpm in an Eppendorf centrifuge.
Before proceeding to immunoisolating the compartment of choice, a prepurification step can be useful. A simple method for partially purifying organelles is density centrifugation, e.g., on a sucrose gradient. Floatation gradients usually give purer fractions, but sedimentation gradients also work well, in most cases. As our compartment of choice (the recycling endosome of MDCK cells) does not float well out of a high-density sucrose solution, we use a sedimentation gradient.
Steps
- Form a step gradient in an SW60 ultraclear tube (Cat. No. 344062; Beckman Coulter, Inc.) by overlaying 1 ml of 20% sucrose (216.2 g/liter sucrose, 3 mM immidazole, pH 7.4) on top of 2.5ml of 35% sucrose (403.0 g/liter sucrose, 3 mM immidazole, pH 7.4). Then add 500 µl of the precleared PNS on top of the gradient.
- Centrifuge for 1 h at 35,000 rpm in an SW60 rotor.
- Discard the upper band, which is often very faint. Use a 200-µl pipette to collect the lower band (usually well visible), which contains endosomes and many other membranes. Cut off about 0.5cm of the yellow tip to avoid breaking the endosomes, which are fragile at this high sucrose density.
Steps
- Preclear the antibody and the PNS for 5 min at 13,000rpm in an Eppendorf centrifuge.
- Add 15µl of 9E10 culture supernatant (2mg/ml) to 500µl of PNS (4mg/ml in HB+). Rotate in a cold room at 2 rpm for 45 min. Add 50 µl of 3 M KCl. Rotate for another 15 min.
- Apply to a sucrose step gradient as described earlier, centrifuge, and retrieve the lower band.
- Measure the protein concentration and use 25 µg per point for immunoisolation.
- Use 50 µl bead slurry per point (2 × 107 beads) for the immunoisolation. Prepare the beads by washing three times with PBS/bovine serum albumin (BSA) (5mg/ml, 0.45µm filtered) in a 15-ml Falcon tube by centrifugation. Take the final bead pellet up in 100µl PBS/BSA per point.
- For each immunoisolation point, add 25µg of membranes to an Eppendorf tube and complete to 900µl with PBS/BSA. Keep enough endosomal fraction for Western blots (see step 3 of the protocol for sample analysis).
- Preclear 5min at 13,000rpm and transfer the supernatants to new tubes.
- Add 100µl of bead slurry to each Eppendorf tube. Be very careful to distribute the beads equally.
- Rotate for 2 h at 2rpm in the cold room.
- Retrieve the beads with a magnet. Keep the supernatants (see step 2 of the protocol for sample analysis).
- Wash with 1 ml of PBS/BSA/O.3M KCl, with 1 ml PBS/BSA, and finally with 1 ml PBS. For all washes, retrieve the beads with the magnet. Do not resuspend with a pipette as this could cause shearing of the endosomes. Instead, add the washing solution and resuspend by rotating (2 rpm) for 2 min on a wheel in the cold room. After the last wash, do not use the magnet, but pellet at 13,000rpm. This allows one to remove the last drop of liquid so that the sample buffer is not diluted.
- It is possible to include a carbonate wash: take up the initial bead pellet in 1 ml 0.1M carbonate buffer, pH 11.0, and rotate for 15 min on the wheel. Then wash as just described.
Steps
- For analysis on minigels, resuspend the bead pellets in 30µl of 1 × sample buffer. You can load the beads with the sample buffer into the wells as, unlike traces of salt or BSA, they do not perturb the migration.
- Pellet the supernatants (unbound material) for 20min at 200,000g in a TLA100.2 rotor. Take up the pellets directly in sample buffer.
- Dilute different amounts (e.g., 10, 25, 50%) of the starting material to 1 ml with PBS/BSA and pellet for 20min at 200,000g in a TLA100.2 rotor. Take up the pellets directly in sample buffer.
- Load the pelleted starting material and the pelleted unbound material on the gel next to the immunoisolated fractions to calculate the yield by comparison (Fig. 1). Always include a negative control (see Section IV), as unspecific stickage may vary considerably from time to time depending on the state of aggregation of the starting material and on the quality of the antibody preparation.
- Proteins that localize on the same compartment as the antigen used for immunoisolation will be isolated with a similar yield as the antigen itself, whereas proteins that only partially colocalize will be isolated with inferior yields (Fig. 2).
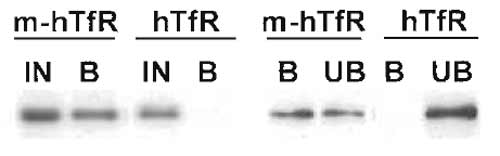 |
| FIGURE 1 Immunoisolation of recycling endosomes. Transferrin receptor-positive endosomes were isolated as described, and fractions were analysed by Western blotting for the presence of the transferrin receptor. Yields were calculated by two means. (Left) The bound fraction (B) is compared with 50% of the starting material (IN). (Right) The bound fraction (B) is compared with the unbound fraction (UB). Both comparisons indicate a yield around 50%. In both types of analysis, a negative control is included consisting of fractions obtained in an identical manner from cells that do not contain the antigen for immunoisolation (a myc tag). |
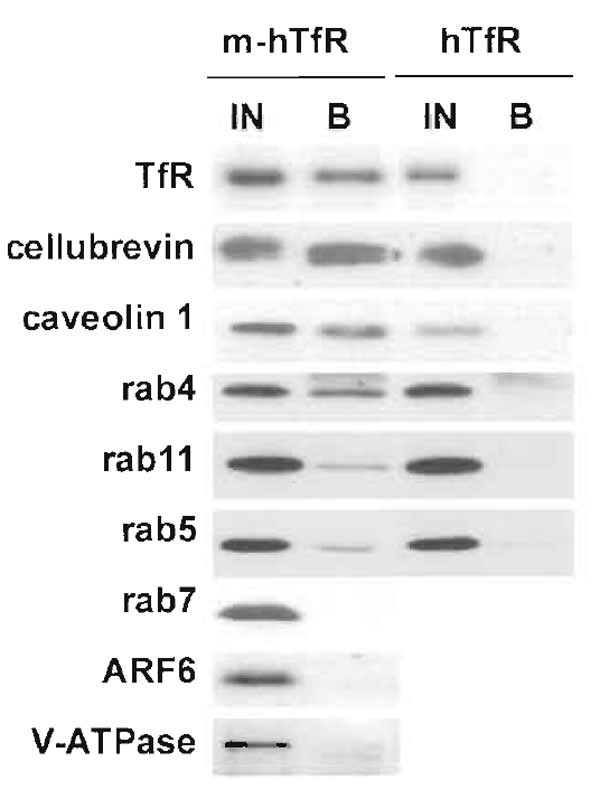 |
| FIGURE 2 Immunoisolation of recycling endosomes. Transferrin receptor-positive endosomes were isolated as described, and fractions were analysed by Western blotting for the presence of various proteins. To estimate the yield of the immunoisolation and the degree of colocalization, bound fractions were compared with 50% of the starting material. Some markers, such as cellubrevin or caveolin 1, were isolated with similar yields as the transferrin receptor itself, indicating an excellent colocalization. Others, such as rab4, rab5, or rab11, were isolated to a lesser extent, indicating only partial colocalization. Finally, some proteins, including rab7, ARF6, and the 110-kDa subunit of the vacuolar ATPase, were absent, indicating that they do not localize to recycling endosomes. |
On the Importance of Negative Controls
Immunoisolation is a powerful technique for analyzing the molecular composition of organelles. However, you can be easily misled by false positives if unspecific binding is not assessed accurately.
Whereas the components of the compartment of choice are isolated with similar yields in different experiments, contaminants are present in different proportions (Fig. 3). So to be confident that a given molecule is present in the isolated compartment, you should check that it is always isolated with similar efficiency with respect to the antigen used for immunoisolation.
In the absence of an appropriate negative control, you can use internal controls, provided that enough is known about the composition of the isolated compartment. Indeed, if several molecules that are known to be absent from the compartment are not isolated whereas several known components are isolated, you can be fairly confident that the immunoisolation is specific.
Due to its abundance in the cell, the endoplasmic reticulum is a frequent contaminant during subcellular fractionation so it may be useful to check that its known components, e.g., the transmembrane protein calnexin, are absent from your isolated fractions (Fig. 3).
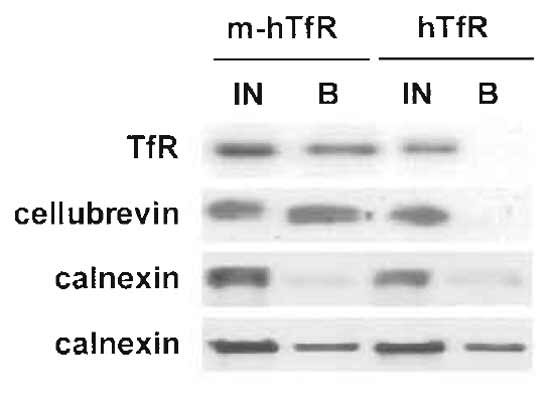 |
| FIGURE 3 Immunoisolation of recycling endosomes. Transferrin receptor-positive endosomes were isolated as described, and fractions were analysed by Western blotting for the presence of contaminants. The ER marker calnexin is isolated with variable yields, but is always also present in the negative control (fractions from cells without the myc epitope). The transferrin receptor and cellubrevin, however, are isolated with a 50% yield in each experiment (only one experiment is shown here) and are not present in the negative control sample. |
We find that unspecific binding correlates best with the quality of the antibody preparation used for immunoisolation. Hence, a negative control should directly test the state of the antibody. In our experiments, this is achieved by performing all experiments in parallel with cells that express the myc-tagged human transferrin receptor and cells that express the untagged receptor. Thus, the negative control sample contains both cellular membranes and the antibody; it lacks only the 13 amino acid myc epitope.
It is important to keep in mind that some antigen-antibody pairs just do not work for immunoisolation, either because the affinity is not high enough or because the antigen is not well exposed. If, after the first attempts, the yield is too low or the background is too high, you can try to optimize the conditions, e.g., by varying the amount of antibody, membranes, or beads. However, if you do not observe any improvement, it might be wise to simply drop this particular antigen-antibody combination and try another antibody or even another component of the compartment as the antigen for immunoisolation.
References
Gagescu, R., Demaurex, N., Parton, R. G., Hunziker, W., Huber, L. A., and Gruenberg, J. (2000). The recycling endosome of Madin- Darby canine kidney cells is a mildly acidic compartment rich in raft components. Mol. Biol. Cell 11, 2775-2791.
Gruenberg, J. (2001). The endocytic pathway: A mosaic of domains. Nature Rev. Mol. Cell Biol. 2, 721-730.
Howell, K. E., Devaney, E., and Gruenberg, J. (1989). Subcellular fraceionation of tissue culture cells. Trends Biochem. Sci. 14, 44- 47.
Howell, K. E., Schmid, R., Ugelsead, J., and Gruenberg, J. (1989). Immunoisolation using magnetic solid supports: Subcellular fractionation for cell-free functional studies. In "Methods in Cell Biology" (A. M. Taratakoff, ed.), pp. 265-292, Academic Press, New York.
Jones, S. M., Dahl, R. H., Ugelstad, J., and Howell, K. E. (1998). Immunoisolation of organelles using magnetic solid supports. In "Cell Biology: A Laboratory Handbook" (J. Celis, ed.), 2nd Ed., pp. 12-25. Academic Press, New York.
Support our developers




