Isolation of Rough and Smooth Membrane Domains of the Endoplasmic Reticulum from Rat Liver
Subcellular fractionation from tissue homogenates have contributed extensively to our understanding of organelle structure and function. This article provides details of a tissue fractionation protocol evolved from several earlier methods (Paiement et al., 1980; Paiement and Bergeron, 1983; Lavoie et al., 1996), for the purification of rough microsomes, corresponding to derivatives of the rough endoplasmic reticulum, and for the purification of smooth microsomes, corresponding to derivatives of the smooth endoplasmic reticulum. The latter constitute a subcompartment of the transitional endoplasmic reticulum of hepatocytes (Lavoie et al., 1996, 1999; Roy et al., 2000).
II. MATERIALS AND INSTRUMENTATION
Sucrose solutions (0.25, 0.86, 1.0, 1.38, and 2.0M solutions needed) 1
1 Densities of sucrose stock solutions should be checked for proper density with a refractometer.
Sucrose-Imidazole (0.25 mM sucrose, 3 mM imidazole, pH 7.4)
100-ml Beaker
Lint-free tissues (e.g., Kimwipes)
Surgical scissors
Potter-Elvehjem tissue grinder with ribbed Teflon pestle (50-ml capacity)
Nylon mesh (150µm mesh, Thompson B & SH Co. Ltd., 8148 Devonshire, Montreal, Canada)
Glass funnel (60° funnel angle, 100mm diameter)
100-ml graduated cylinder
Refrigerated low-speed centrifuge with fixed angle rotor (e.g., Beckman-Coulter Avanti J-20 with JA 25.50 rotor or equivalent)
Pasteur Pipettes
Loose-fitting Dounce homogenizer (7- and 15-ml capacity, with Wheaton type B piston, at least two of each)
Graduated conical tube
Parafilm
2.0-ml glass syringes with blunt-end syringe needles (10 cm long, 2 mm diameter)
Ultracentrifuge with swinging bucket and fixed-angle rotors (e.g., Beckman L-70 with SW60 and Ti50 rotors)
- Rats must be fasting for 12 to 24h prior to sacrifice.
- Use two male rats of 300- to 400-g body weight (one liver weighs approximately 9 g).
- Sacrifice rats via decapitation 2 and excise livers (free of connective tissue) and place in petri dish containing ice-cold 0.25M sucrose.
2 Animal sacrifice must be carried out according to the "Guide to the Care and Use of Experimental Animals of the Canadian Council on Animal Care" or equivalent protocol required by the country concerned. - Blot the livers using lint-free tissues and transfer to a preweighed beaker containing ice-cold 0.25M sucrose and weigh (weight of two livers together should be a minimum of 18 g).
- Blot the livers using lint-free tissues and chop them finely with scissors. Allow the pieces to fall into the mortar of the tissue grinder. 3
3 Steps 5 to 8 are carried out in a cold room. - Add a volume of ice-cold 0.25M sucrose, which is equivalent to 2.5 times the weight of the livers (e.g., 18.0 g × 2.5 = 45.0 ml).
- Homogenize using the ribbed Teflon pestle for 20 strokes (down and up is one stroke) at 2850rpm (mortar should be kept in plastic beaker filled with ice throughout homogenization process).
- Filter the homogenate through the nylon mesh folded over four times in the glass funnel. 4
4 The homogenate should not be squeezed through the nylon mesh, but instead stirred gently with a glass rod to facilitate the homogenate passing through the mesh. - The total homogenate at this point should be approximately 54ml (45ml sucrose plus about 9ml liver tissue) and should be separated into 4 × 15ml tubes.
- Centrifuge in a Beckman-Coulter JA 25.50 fixed angle rotor that is precooled to 4°C at 8700g for 13 min to sediment cellular debris and nuclei.
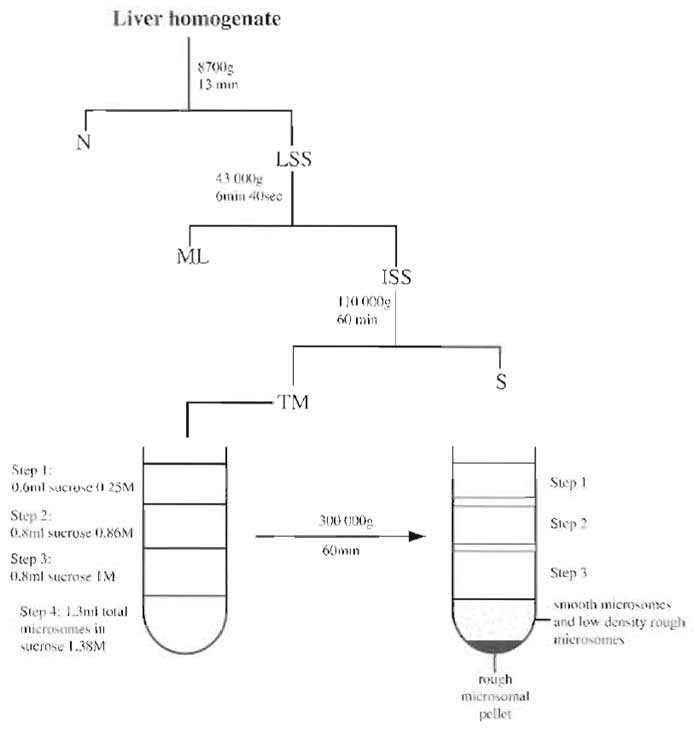
- Transfer the supernatant [low-speed supernatant (LSS fraction), see Fig. 1] into three polycarbonate screw top tubes (10-ml capacity).
- Centrifuge in a Ti50 fixed angle rotor at 43,000g for 6 min 40 s [press the stop button of the centrifuge at exactly 6min 40s. Complete stop (with brakes) should occur by about 10 min 20 s after the start of centrifugation].
- With a Pasteur pipette, recover the supernatant [intermediate-speed supernatant (ISS fraction), see Fig. 1] and a portion of the flocculent layer that is covering the pellet (ML fraction, containing lysosomes and mitochondria).
- Centrifuge the ISS fraction in a Ti50 rotor at 110,000 g for 60 min.
- Discard supernatant (S fraction, see Fig. 1).
- Resuspend the pellets in 1.38 M sucrose. 1 Recovery of total microsomes (TM) in the pellets is done as follows.
- Add 0.5ml of 2.0M sucrose1 to the first centrifuge tube and resuspend the microsomal pellet using a blunt-ended glass rod.
- Transfer the suspension to subsequent tubes, resuspending the microsomal pellet with the glass rod in each one before transferring on to the next.
- After resuspension is complete, rinse all centrifuge tubes with 2.0M sucrose and transfer to a 7-ml Dounce.
- Repeat rinsing as needed. However, do not exceed an accumulation of 5ml in the Dounce.
- Gently homogenize the resuspensions in the Dounce using the piston.
- Measure volume using a graduated conical tube.
- Rinse both the centrifugation tubes and the Dounce with 1 ml 2.0M sucrose, 1 which is transferred to the conical tube and mixed in gently by covering the top of the tube with parafilm and inverting the tube two or three times. Repeat once.
- Use a refractometer to adjust the density to 1.38M by adding 0.25 or 2.0M sucrose1 to the TM resuspension as needed.
- Bring the volume of the TM resuspension to 9.8 ml using the 1.38 M sucrose1 stock.
- Prepare six sucrose gradients using 4.0-ml tubes destined for the Beckman SW60 rotor as follows.
- Carefully place 0.6ml of 0.25M sucrose1 in the bottom of a Beckman SW60 tube with a glass syringe and blunt-ended needle.
- Using a new syringe and needle, gently place the needle at the bottom of the tube and very gently inject 0.8 ml of 0.86M sucrose1 underneath previous layer.
- Place a third syringe and needle carefully at the bottom of the tube and inject 0.8ml of 1.0M sucrose1 very gently underneath the previous two layers.
- Using a fourth new syringe and needle, very gently insert needle to the bottom of the tube again and very carefully inject 1.4ml of TM fraction to produce the bottom layer of the gradient (see Fig. 1).
- Repeat steps a-e for all six tubes.
- Centrifuge gradients in a SW60 swingingbucket rotor at 53,000rpm (300,000g) for 60min.
- For smooth microsomes (SM):
- Carefully extract the top half of the bottom step (see step 4 in Fig. 1) using a Pasteur pipette and place in a Ti50 centrifuge tube.
- Add 0.25M sucrose-imidazole until tube is filled and mix gently.
- For rough microsomes (RM):
- Discard all gradient steps in all tubes.
- Turn tubes upside down on a lint-free tissue for approximately 30s to allow residual supernatant to drip out.
- Wipe edges of tube (above pellet) using a lint-free tissue wrapped around a glass rod.
- Resuspend pellets using a 15.0-ml Dounce and rinse tubes with sucrose-imidazole (using the same sequential rinsing method as described in Steps 16a to 16e). Transfer resuspended rough microsomes to a Ti50 tube.
- Once sequential rinsing is done, add sucrose-imidazole to tube so that the final volume of sucrose-imidazole added is equivalent to approximately 11 ml.
- Divide both SM and RM separately into two equal portions in Beckman Ti50 tubes and centrifuge at 140,000g for 60 min.
- Discard supernatants and resuspend as follow:
- Resuspend the rough microsomal pellet in 2.0 ml sucrose-imidazole.
- Resuspend the smooth microsomal pellet in 3.0 ml sucrose-imidazole.
- Aliquot microsomal fractions into 60-, 110-, and 210-µl portions and store at -80°C in hermetically sealed tubes until needed.
 |
| FIGURE 1 |
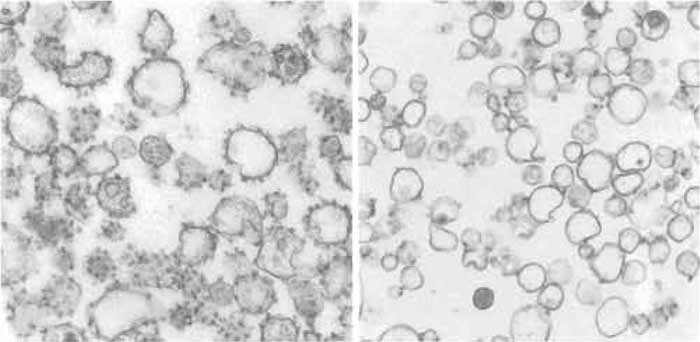
The "rough" nature of the derivatives of the rough endoplasmic reticulum can be confirmed by examination of preparations by electron microscopy (Fig. 2). Samples were fixed in suspension and processed as described previously (Paiement et al., 1980). Classical rough microsomes are shown in Fig. 2A and contain greater than 80% rough vesicles as determined by quantification of the number of vesicles with associated ribosomes. In Fig. 2B the microsomal fraction consists of approximately 60% smooth microsomal vesicles and 40% rough microsomal vesicles, which have few (one to four) ribosomes associated with their limiting membranes. Both microsomal fractions were shown previously to be enriched in enzyme markers for the endoplasmic reticulum and reduced in enzyme markers for Golgi and plasma membrane derivatives (Paiement et al., 1980; Paiement and Bergeron, 1983; Lavoie et al., 1996, 1999; Roy et al., 2000).
 |
| FIGURE 2 |
This procedure takes approximately 12h. All samples should be kept on ice at all times throughout this protocol. All centrifugations should take place at 4°C.
References
Lavoie, C., Lanoix, J., Kan, F. W. K., and Paiement, J. (1996). J. Cell Sci. 109, 1415-1425.
Lavoie, C., Paiement, Dominguez, M. J., Roy, L., Dahan, S., Gushue, J. N., and Bergeron, J. J. M. (1999). J. Cell Biol. 146, 285-299.
Paiement, J., and Bergeron, J. J. M. (1983). J. Cell Biol. 96, 1791-1796.
Paiement, J., Beaufay, H., and Godelaine, D. (1980). J. Cell Biol. 86, 29-37.
Roy, L., Bergeron, J. J. M., Lavoie, C., Hendriks, R., Gushue, J., Fazel, A., Pelletier, A., Morr6, D. J., Subramaniam, V. N., Hong, W., and Paiement, J. (2000). Mol. Biol. Cell. 11, 2529-2542.
Support our developers




