Preparation of Synaptic Vesicles from Mammalian Brain
Synaptic vesicles are secretory organelles that store neurotransmitters in presynaptic nerve endings. When an action potential arrives in the nerve terminal, the plasma membrane is depolarized, leading to the opening of voltage-gated Ca2+ channels. The rise in intracellular Ca2+ concentration leads to exocytosis of synaptic vesicles within a time interval that can be as short as 200 µs (reviewed by Südhof, 1995).
Synaptic vesicles possess several remarkable properties that distinguish them from most other organelles involved in membrane traffic. First, they are very abundant in brain tissue. Model calculations show that an average neuron contains approximately 106 synaptic vesicles, with a total of around 1017 in the human central nervous system (Jahn and Sfidhof, 1993). Approximately 5% of the protein in the brain is contributed by synaptic vesicles; thus, about a 20-fold enrichment from homogenate is sufficient to obtain a pure preparation. Second, synaptic vesicles are highly homogeneous in size and shape and, in addition, are smaller than most other organelles, with an average diameter of only 50nm. Therefore, size-fractionation techniques can be applied for the isolation of synaptic vesicles. Third, synaptic vesicles do not contain a matrix of soluble proteins (Jahn and Sfidhof, 1993), as they recycle many times in the nerve terminal and thus can only be reloaded with nonpeptide transmitters by means of specific transport systems.
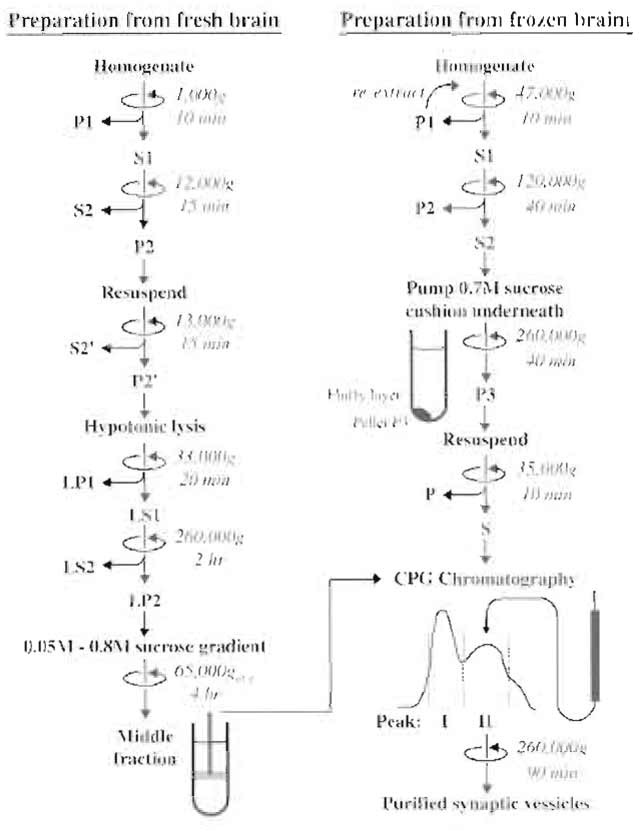 |
| FIGURE 1 Flowchart depicting the main steps of the two preparation methods for synaptic vesicles described in the text. The final step for both methods is CPG chromatography. |
The following chemicals are used: HEPES (Research Products International, #H75030), sucrose (Sigma, # S-9378), glycine (Bio-Rad, 161-0718), phenylmethylsulfonyl fluoride (PMSF; ICN, #195381), pepstatin A (ICN, #195368), dimethyl sulfoxide (DMSO, Sigma, #D-8779), and controlled pore glass beads (CPG Inc., see Appendix). Note that the standard reagents can also be obtained from other sources.
The following instrumentation is required: loosefitting, motor-driven glass-Teflon homogenizer (Braun, Melsungen, Germany), cooled centrifuge [Sorvall RC5 (DuPont) or comparable, SS34 rotor], ultracentrifuge with fixed angle and swing-out rotors [Beckman L80 (Beckman Instruments) or comparable, Ti70 or Ti50.2 rotor, SW-28 rotor, Ti 45 rotor] and corresponding tubes, equipment for column chromatography (peristaltic pump, UV monitor, fraction collector), gradient mixer for forming continuous sucrose gradients, filtration device for the filtration of buffers using 0.45-µ membranes (Millipore), and glass columns (see Appendix).
A. Preparation of Synaptic Vesicles from Synaptosomes
In this protocol (Nagy et al., 1976; Huttner et al., 1983), a crude synaptosomal fraction (P2) is first isolated by differential centrifugation. The synaptosomes are then lysed by osmotic shock and synaptic vesicles are released into the medium. After removal of synaptosomal fragments and large membranes, synaptic vesicles are sedimented by high-speed centrifugation. The resulting pellet, already five- to sixfold enriched in synaptic vesicles, is then purified further by sucrose velocity-density gradient centrifugation and sizeexclusion chromatography on controlled pore glass beads (CPG). This procedure is the standard method for obtaining synaptic vesicles of the highest purity, with less than 5% contamination as judged by electron microscopy and biochemical analysis. This preparation does contain, however, endosomes derived from nerve terminals and decoated coated vesicles that lost their clathrin but retained adaptors. The degree of this contamination is probably minor but cannot be quantified easily.
Solutions
- Homogenization buffer: 320 mM sucrose, 4 mM HEPES-NaOH, pH 7.3 (HEPES is optional; we found no difference when the buffer is omitted. Other buffers such as MES and slightly lower pH are also acceptable).
- 1 M HEPES-NaOH, pH 7.4
- 40 mM sucrose
- 50 mM sucrose
- 800 mM sucrose
- Glycine buffer: 300 mM glycine, 5 mM HEPES-KOH, pH 7.4, degassed and filtered through a 0.45-µm filter
- Protease inhibitors: 1 mg/ml pepstatin A in DMSO and 200 mM PMSF in dry ethanol. Keep stocks at room temperature. Add 1/1000 volume where indicated. Note that PMSF is unstable in aqueous solutions and should not be added to buffers prior to use. The PMSF stock solution should be prepared fresh every day.
After collecting the brains, all steps are carried out on ice or at 4°C.
- Decapitate 20 rats (about 2 months old; 180- 200g body weight), remove the brains, avoiding myelin-rich areas such as corpus callosum or medulla oblongata, place into 180ml ice-cold homogenization buffer, and homogenize in several aliquots with a loose-fitting glass Teflon homogenizer (nine strokes, 900rpm). Add protease inhibitors.
- Centrifuge the homogenate for 10min at 1000gmax (2700rpm, Sorval SS34 rotor), discard the resulting pellet (P1) containing large cell fragments and nuclei, and collect the supernatant (S1).
- Centrifuge S1 for 15min at 12,000gmax (10,000rpm; SS34 rotor); remove the supernatant (S2) containing small cell fragments such as microsomes or small myelin fragments and soluble proteins. Wash the pellet (P2) by carefully resuspending in 120ml homogenization buffer (pipette, avoiding the dark brown bottom part of the pellet that consists mainly of mitochondria) and recentrifuging at 13,000gmax (11,000rpm, SS34 rotor); discard the supernatant (S2'). The resulting pellet (P2') represents a crude synaptosomal fraction.
- To release synaptic vesicles from the synaptosomes, resuspend P2' in homogenization buffer to yield a final volume of 12 ml. Transfer this fraction into a glass-Teflon homogenizer, add 9 volumes (108ml) ice-cold water and perform three up-and-down strokes at 2000rpm. Add 1 ml of 1M HEPES-NaOH, pH 7.4, and protease inhibitors.
- Centrifuge the suspension for 20min at 33,000gmax (16,500rpm, SS34 rotor) to yield the lysate pellet (LP1) and the lysate supernatant (LS1). Using an electric pipetter, carefully remove LS1 immediately after the end of the run without disturbing LP1. It is crucial that LS1 does not get contaminated even with traces of membrane fragments from LP1 (rather, leave 1-2ml behind in the tube). Contaminating LS1 with LP1 is the most common problem, which significantly reduces the purity of the final vesicle fraction.
- Centrifuge LS1 for 2h at 260,000gmax (50,000rpm, Beckman 60Ti or comparable rotor). Discard the supernatant (LS2) and resuspend the pellet (LP2) in 6 ml of 40mM sucrose utilizing a small, tightfitting glass-Teflon homogenizer. Extrude the resuspended sample consecutively through a 23- and a 27-gauge hypodermic needle attached to a 10-ml syringe (avoid air bubbles).
- Layer the suspension (3-ml aliquots) on top of a linear sucrose gradient formed from 18.5ml of 800 mM sucrose and 18.5 ml of 50 mM sucrose (prepare two tubes containing identical gradients in advance) and centrifuge for 4h at 65,000gav (25,000rpm, Beckman SW 28 rotor). After the run, a turbid (whiteopaque) zone is visible in the middle of the gradient (in the range of 200 to 400 mM sucrose, best seen when viewed against a black background with light from the top). Collect these bands with the aid of a glass capillary connected to a peristaltic pump, yielding a combined volume of 25-30ml. This fraction represents synaptic vesicles that are 8- to 10-fold enriched over the homogenate (Jahn et al., 1985). Note that synaptic vesicles do not reach isopycnic equilibrium during this velocity gradient-type centrifugation. Changes of angular velocity or of the run time will therefore affect the result.
- Equilibrate a CPG-3000 column (180 × 2cm, see Appendix) with 10 column volumes of glycine buffer (optimally done overnight before the preparation). Load the sample on top of the resin and overlay it carefully with glycine buffer without diluting the sample. Elute the column with glycine buffer at a flow rate of 40ml/h, collecting 6- to 8-ml fractions. Monitor protein effiux at 280 nm. The first peak contains plasma membranes and some microsomes and is usually smaller than the second peak containing synaptic vesicles. If no separation into two clearly distinguishable peaks is obtained, the column may need to be repacked. Fractions of the second peak are pooled and centrifuged for 90min at 260,000gmax (50,000rpm, Beckman 60Ti rotor). The synaptic vesicle pellet should have a glassy appearance, being completely transparent and colorless. Resuspend it in the desired buffer as in step 6. The suspension is frozen rapidly (e.g., in liquid nitrogen) and stored at -70°C. Yields are typically between 2 and 3 mg of protein, based on one of the commercially available Coomassie blue protein determination kits.
B. Preparation of Synaptic Vesicles from Frozen Brain
This procedure starts with a harsh homogenization of frozen brains to efficiently break up the nerve terminals, thus releasing synaptic vesicles. Frozen brains are ground in a precooled mortar to yield a fine powder. This treatment does not affect the function or integrity of the small synaptic vesicles, but larger membrane structures are ruptured. After resuspending the tissue powder in sucrose solution, most of the cell fragments are removed by centrifugation with low and intermediate angular velocities, leaving synaptic vesicles in the supernatant. Synaptic vesicles are then sedimented at high speed through a cushion of 0.7M sucrose, removing soluble proteins and membrane contaminants of lower buoyant density (mostly myelin). Synaptic vesicles are five- to sixfold enriched in the pellet and can be purified further by CPG chromatography.
Solutions
- Homogenization buffer: 320mM sucrose, degassed
- 700mM sucrose: 700mM sucrose and 10mM HEPES-KOH, pH 7.3
- Resuspension buffer: 320 mM sucrose and 10 mM HEPES-KOH, pH 7.3
- Glycine buffer: 300mM glycine and 5mM HEPES-KOH, pH 7.3, degassed
- Protease inhibitors: 1 mg/ml pepstatin A dissolved in DMSO and 200mM PMSF in dry ethanol; add 1 / 1000 volume where indicated
After powdering the frozen brains, all steps are carried out on ice or at 4°C.
- Decapitate 40 rats (2 months old, 180-200 g body weight); remove the brains, avoiding myelin-rich areas such as corpus callosum or medulla oblongata, and freeze immediately in liquid nitrogen. Immediate shock freezing is essential. In our experience, frozen brains available from commercial sources are usually not satisfactory for this reason.
- To create a tissue powder, place the frozen brains into a porcelain mortar precooled with liquid nitrogen. Cover them with cheesecloth and break them carefully using a porcelain pestle. Grind to a fine powder. This step is crucial for obtaining high yields. After evaporation of the liquid N2, suspend the powder in 320ml ice-cold homogenization buffer (magnetic stirrer) and homogenize with a glass-Teflon homogenizer (eight strokes, 1000 rpm).
- Centrifuge the homogenate for 10min at 47,000gmax (20,000rpm, Sorval SS-34 rotor). Collect the supernatant (S1). The pellet (P1) contains large cell fragments and nuclei, but also some entrapped synaptic vesicles. To increase the yield, reextract the pellet with 160ml homogenization buffer by means of one slow stroke in the glass-Teflon homogenizer followed by centrifugation as described earlier. The resulting supernatant (S1') is combined with S1.
- Centrifuge S1 for 40min at 120,000gmax (32,000rpm, Beckman 45Ti rotor). Using an electric pipetter, collect the supernatant (S2) carefully without disturbing the pellet (P2). It is crucial that S2 is not contaminated with membrane fragments from the soft pellet P2. S2 should be clear with a reddish color. If it is turbid, it should be recentrifuged using the same conditions to remove contaminating membrane fragments.
- To sediment synaptic vesicles through a sucrose cushion, fill 25-ml centrifuge tubes appropriate for a Beckman 60Ti rotor with 20ml S2. Form the sucrose cushion by pumping 5.5 ml of 700mM sucrose underneath S2 using a peristaltic pump and a glass capillary. Centrifuge for 2h at 260,000gmax (50,000rpm, Beckman 60Ti rotor). Remove the supernatant S3 and resuspend the pellet P3 in 6-10 ml resuspension buffer with a small, tight-fitting glass-Teflon homogenizer. Extrude the resuspended sample consecutively through a 23- and a 27-gauge hypodermic needle attached to a 10-ml syringe (avoid air bubbles). This sample represents a crude synaptic vesicles fraction. Clear the suspension by a short spin (10min) at 35,000gmax (17,000 rpm, SS34) before loading onto the CPG-column.
- Equilibrate a CPG column (see Appendix) with 10 column volumes of glycine buffer. Load the sample on top of the resin and overlay carefully with glycine buffer without disturbing the sample. A column of size 85 × 1.6 cm has a maximal capacity of 15 mg of protein, requiring several consecutive runs if all material is to be chromatographed. Elute the column with glycine buffer at a flow rate of 80ml/h, collecting 2-ml fractions. Follow the elution of protein with a UV detector at 280nm. The first peak, containing plasma membranes and microsomes, is usually larger than the second peak, containing synaptic vesicles. The two peaks are typically not completely separated in this protocol. The shoulder frequently observed at the end of the second peak represents soluble protein. Pool the fractions of the second peak and centrifuge for 2h at 260,000gmax (50,000rpm; 60Ti rotor). Resuspend the synaptic vesicle pellet in the desired buffer as described in step 5.
Scaling up or down is feasible but it should be kept in mind that changing rotors or using half-filled centrifuge tubes may affect yield and purity significantly and adversely. Contamination by other subcellular fractions (e.g., plasma membranes, mitochondria, endoplasmic reticulum) can be monitored conveniently by assaying for marker enzymes (Hell et al., 1988). In parallel to a decrease of these marker enzymes, proteins specific for synaptic vesicles, namely synaptophysin (p38), for which antibodies are available commercially (e.g., from Boehringer, Mannheim, Germany), should be enriched about 20- to 25-fold over homogenate (Jahn et al., 1985), best quantitated by immunoblotting.
Synaptobrevin is less reliable for quantification by SDS-PAGE/immunoblotting as histones present in the homogenate migrate alongside synaptobrevin/VAMP in nuclei-containing fractions (homogenate) and interfere with the signal, resulting in overestimation of the enrichment factor. The protein profile of the synaptic vesicles preparation as observed after SDS-PAGE exhibits a characteristic pattern (Huttner et al., 1983; Hell et al., 1988), with the prominent membrane proteins synaptobrevin/VAMP, synaptophysin (p38), synaptotagmin (p65), and synapsin I being clearly visible. Synaptic vesicle preparations contain various amounts of soluble proteins with affinity for membranes such as glyceraldehyde phosphate dehydrogenase, aldolase, actin, and tubulin.
The morphology of the synaptic vesicles fraction can be studied using electron microscopy (Fig. 2). Membranes can be visualized easily, e.g., by negative staining (Hell et al., 1988). Synaptic vesicle membranes are identified by their very uniform appearance (small vesicular profiles of approximately 50nm diameter). Confirmation can also be obtained by immunogold labeling for the vesicle protein synaptophysin, which can be carried out conveniently on a single day when combined with negative staining (Jahn and Maycox, 1988).
 |
| FIGURE 2 Electron micrograph showing a synaptic vesicle fraction purified by the procedure described in Section IIIB (negative staining). (Inset) Magnification of a field following immunogold labeling for the synaptic vesicle protein synaptophysin. For methods, see Jahn and Maycox (1988). Bar: 200nm. Electron micrographs courtesy of Dr. Peter R. Maycox (London, UK). |
The authors thank Dr. Duane D. Hall for preparation of Fig. 1.
References
Burger, R M., Mehl, E., Cameron, R, Maycox, R R., Baumert, M., Lottspeich, E, De Camilli, P., and Jahn, R. (1989). Synaptic vesicles immunoisolated from rat cerebral cortex contain high levels of glutamate. Neuron 3, 715-720.
Hell, J. W., Maycox, P. R., Stadler, H., and Jahn, R. (1988). Uptake of GABA by rat brain synaptic vesicles isolated by a new procedure. EMBO J. 7, 3023-3029.
Huttner, W. B., Schiebler, W., Greengard, P., and De Camilli, P. (1983). Synapsin I (protein I), a nerve terminal-specific phosphoprotein. III. Its association with synaptic vesicles studied in a highly purified synaptic vesicle preparation. J. Cell Biol. 96, 1374- 1388.
Jahn, R., and Maycox, P. R. (1988). Protein components and neurotransmitter uptake in brain synaptic vesicles. In "Molecular Mechanisms in Secretion" (N. A. Thorn, M. Treiman, and O. H. Peterson, eds.), pp. 411-424. Munksgaard, Copenhagen.
Jahn, R., Schiebler, W., Ouimet, C., and Greengard, P. (1985). A 38,000 dalton membrane protein (p38) present in synaptic vesicles. Proc. Natl. Acad. Sci. USA 82, 4137-4141.
Nagy, A., Baker, R. R., Morris, S. J., and Whittaker, V. P. (1976). The preparation and characterization of synaptic vesicles of high purity. Brain Res. 109, 285-309.
Reimer, R. J., Fremeau, R. T., Jr., Bellocchio, E. E., and Edwards, R. H. (2001). The essence of excitation. Curr. Opin. Biol. 105, 273- 281.
Südhof, T. C. (1995). The synaptic vesicle cycle: A cascade or proteinprotein interactions. Nature 375, 645-653.
Walch-Solimena, C., Takei, K., Marek, K., Midyett, K., Siidhof, T. C., De Camilli, P., and Jahn, R. (1993). Synaptotagmin: A membrane constituent of neuropeptide-containing large dense-core vesicles. J. Neurosci. 13, 3895-3903.
Support our developers




