Preparation of Proteasomes
The proteasome is a protein-destroying machine capable of degrading a variety of proteins involved in the regulation of diverse processes, such as the cell cycle, immune response, signaling cascades, and developmental programs in various eukaryotes (Hershko and Ciechanover, 1998; Kloetzel, 2001; Rock et al., 2002). Early studies identified the proteasome as a latent protease complex with a sedimentation coefficient of 20S, and it was accordingly named the 20S proteasome (Coux et al., 1996). The 20S proteasome is a barrel-like particle formed by the axial stacking of four rings made up of two outer α rings and two innerβ rings, being associated in the order of αββα. The active sites reside in a chamber formed by the centers of the abutting β rings (Bochtler et al., 1999; Unno et al., 2002). The latency of the proteasome may be explained by the tertiary structure, as the center of the α ring is almost closed, preventing penetration of proteins into the inner aspect of the β ring on which the proteolytically active sites are located (Fig. 1). The 20S proteasome possesses a variety of catalytic centers that presumably contribute to the hydrolysis of multiple peptide bonds in single polypeptide substrates by a coordinated mechanism.
 |
| FIGURE 1 Crystal structure of the 20S proteasome from the bovine liver. For details, see Unno et al. (2002). |
Subsequently, the latent 20S proteasome was demonstrated to act as a catalytic core of a large multisubunit proteolytic complex. To date, three protein factors that can stimulate 20S proteasome activity have been described. One is PA700 (also known as the 19S regulatory particle), which can be divided into lid and base complexes. The lid consists of multiple Rpn (non- ATPase) subunits, while the base consists of six proteasomal ATPases, Rptl-Rpt6, and a few additional Rpn subunits (Finley et al., 1998). PA700 can associate with the 20S proteasome in an ATP-dependent manner to form the 26S proteasome with a molecular mass of ~2500 kDa, a eukaryotic ATP-dependent protease (Fig. 2), capable of degrading mainly proteins tagged with a polyubiquitin (Ub) chain, which functions as a degradation signal (Baumeister et al., 1998; Pickart, 2001).
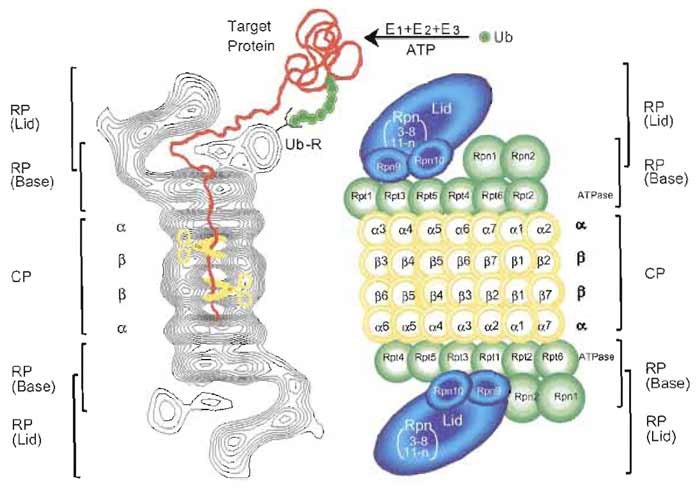 |
| FIGURE 2 Molecular organization of the 26S proteasome. (Left): Averaged image based on electron micrographs of the complex of the 26S proteasome from rat. The α and β rings of the 20S proteasome are indicated. Photograph kindly provided by W. Baumeister. (Right): Schematic drawing of the subunit structure. Ub, ubiquitin; E1 (Ub-activating), E2 (Un-conjugating), and E3 (Ub-ligating) enzymes; CP, core particle; RP, regulatory particle; Rpn, RP non-ATPase; Rpt, RP triple ATPase; Ub-R, Lib receptor (poly-Ub binding subunit). |
The third proteasome activator is PA200, which is localized in the nucleus and is involved in DNA repair (Ustrell et al., 2002).
II. MATERIALS AND INSTRUMENTATION
Q-Sepharose (Cat. No. 17-1014-03), Q-Sepharose fast flow (Cat. No. 17-0510-10), Superdex 200 pg (Cat. No. 17-1043-01), Mono Q (Cat. No. 17-5166-01), and heparin-Sepharose CL-6B (Cat. No. 17-0467-09) can be purchased from Amersham. BiG-Gel A-1.5m (Cat. No. 151-0440) and hydroxylapatite BiG-Gel HTP (Cat. No. 130-0420) are from Bio-Rad. Polyethylene glycol 6000 (Cat. No. P-2139), ubiquitin (Ub, Cat. No. U-6253), succinyl- Leu-Leu-Val-Tyr-4-methyl-courmaryl-7-amide (Suc-LLVY-MCA, Cat. No. S-6510), and Supelco TSKDEAE 650M is from Sigma. Amicon PM-10 and PM-30 membranes (Cat. No. 13132 and 13232) can be obtained from Millipore. Complete protease inhibitor (Cat. No. 1 697 498) is from Roche.
Solutions
- Buffer A: 25 mM Tris-HCl (pH 7.5) containing 1 mM dithiothreitol (DTT) (or 10mM 2-mercaptoethanol) and 20% glycerol
- Buffer B: 10mM phosphate buffer (pH 6.8) containing 1 mM DTT and 20% glycerol
- Buffer C: Buffer A containing 0.5 mM ATP
- Buffer D: Buffer B containing 5 mM ATP
- Buffer E: 10mM Tris-HCl (pH 7.0), 25mM KCl, 10 mM NaCl, 1.1 mM MgCl2, 0.1 mM EDTA, 1 mM DTT, and 10% glycerol
- Buffer F: 10mM Tris-HCl (pH 8.5), 25mM KCl, 10 mM NaCl, 1.1 mM MgCl2, 0.1 mM EDTA, 1 mM DTT, and 10% glycerol
A. Preparation of 20S Proteasomes
Procedures
- Homogenize 200- to 400-g samples of animal tissues in 3 volumes of 25 mM Tris-HCl buffer (pH 7.5) containing 1 mM DTT and 0.25M sucrose in a Potter-Elvehjem homogenizer. Centrifuge the homogenate for 1 h at 70,100g and use the resulting supernatant as the crude extract.
- Add glycerol at a final concentration of 20% to the crude extract. Then mix the extract with 500 g of Q-Sepharose that has been equilibrated with buffer A. Wash the Q-Sepharose with the same buffer on a Biichner funnel and transfer to a column (5 × 60cm). Wash the column with buffer A, elute the material with 2 liters of a linear gradient of 0-0.8 M NaCl in the same buffer, and measure the activity of proteasomes using Suc-LLVY-MCA as a substrate (for details, see Section IV).
- Pool fractions containing 20S proteasomes from the Q-Sepharose column and add 50% polyethylene glycol 6000 (adjust to pH 7.4) at a final concentration of 15 % with gentle stirring. After 15 min, centrifuge the mixture at 10,000g for 20min, dissolve the resulting precipitate in a minimum volume (approximately 50 ml) of buffer A, and centrifuge at 20,000 g for 10 min to remove insoluble material.
- Fractionate the material precipitated with polyethylene glycol on a Bio-Gel A-1.5m column (5 × 90 cm) in buffer A. Collect fractions of 10ml and assay their proteasome activity. Pool fractions of 20S proteasomes.
- Apply the active fractions from the Bio-Gel A- 1.5m column directly to a column of hydroxylapatite equilibrated with buffer B. Wash the column with the same buffer and elute the material with 400ml of a linear gradient of 10-300mM phosphate. Collect fractions of 4ml. Elute the 20S proteasomes with about 150 mM phosphate.
- Combine the active fractions from the hydroxylapatite, dialyze against buffer A, and apply to a column of heparin-Sepharose CL-6B equilibrated with buffer A. Wash the column with the same buffer until the absorbance of the eluate at 280nm returns to baseline. Then eluate with 200ml of a linear gradient of 0-0.4M NaCl in the same buffer and collect fractions of 2ml. Eluate the 20S proteasomes with approximately 75 mM NaCl.
- Pool the fractions with high proteasomal activity, dialyze against buffer A, and concentrate to about 5mg/ml protein by ultrafiltration in an Amicon cell with a PM-10 membrane. The enzyme can be stored at -80°C for at least 2 to 3 years.
Procedures
- Homogenize 200- to 400-g samples of animal tissues in 3 volumes of 25 mM Tris-HCl buffer (pH 7.5) containing 1 mM DTT, 2mM ATP, and 0.25 M sucrose in a Potter-Elvehjem homogenizer. Centrifuge the homogenate for 1 h at 70,100g and use the resulting supernatant as the starting material.
- Recentrifuge the crude supernatant for 5h at 70,100g to obtain 26S proteasomes, which precipitate almost completely. Dissolve the precipitate in a suitable volume (40-50ml) of buffer C and centrifuge at 20,000g for 30min to remove insoluble material.
- Apply samples of the preparation from step 2 to a Bio-Gel A-1.5m column (5 × 90cm) in buffer C. Collect fractions of 10 ml and assay the 26S proteasome activity in the fractions (for the assay, see Section IV). Pool fractions of 26S proteasomes.
- Add ATP at a final concentration of 5 mM to the pooled fractions of 26S proteasomes from the Bio-Gel A-1.5m column. Apply a sample directly to a hydroxylapatite column with a 50-ml bed volume that has been equilibrated with buffer D. Recover the 26S proteasomes in the flow-through fraction because they do not associate with this column in the presence of 5 mM ATP. Approximately 70% of the proteins, including free 20S proteasomes, bind to the hydroxylapatite resin.
- Apply the flow-through fraction from the hydroxylapatite column to a Q-Sepharose column that has been equilibrated with buffer C without ATP and washed with 1 bed volume of buffer C. Wash the column with 5 bed volumes of buffer C and elute the adsorbed materials with 300ml of a linear gradient of 0-0.8M NaCl in the same buffer. Collect fractions of 3.0ml of eluate. Proteins with the ability to degrade Suc-LLVY-MCA with or without 0.05% SDS are eluted with about 0.4M NaCl as a single symmetrical peak. ATPase activity and the ATP-dependent activity necessary to degrade 125I-labeled lysozyme-Ub conjugates are observed at the same position as the peptidase activity and are eluted as superimposable symmetrical peaks, which suggests a specific association of ATPase with the 26S proteasome complex. Collect the protein in fractions exhibiting high activity.
- Concentrate the 26S proteasome fraction obtained by Q-Sepharose chromatography to 2.0mg/ ml by ultrafiltration with an Amicon PM-30 membrane and subject samples of 2.0mg of protein to 10-40% glycerol density gradient centrifugation (30ml in buffer C containing 2 mM ATP). Centrifuge for 22h at 82,200g in a SW rotor and collect 1-ml fractions from the bottom of the centrifuge tube. A single major peak of peptidase activity, obtained in the absence of SDS, is eluted around fraction 15, but when the activity is assayed with 0.05% SDS, another small peak is observed around fraction 20. The latter peak corresponds to the elution position of 20S proteasomes. ATPase activity is observed at the same position as peptidase activity. Activity for ATP-dependent degradation of 125I-labeled lysozyme-Ub conjugates is also observed as a single symmetrical peak, coinciding in position with the ATPase and peptidase activities in the absence of SDS. No significant 125I-labeled lysozyme-Ub conjugate-degrading activity is detected in fractions of 20S proteasomes. Pool fractions 12-16 and store at -80°C.
Steps for homogenization, ultracentrifugation, and Bio-Gel A-1.5m gel filtration are similar to those used for the preparation of 26S proteasomes. Apply the pooled fractions of the 26S proteasome from the Bio- Gel A-1.5m column directly to a hydroxylapatite column with a 50-ml bed volume that has been equilibrated with buffer C. Wash the column with the same buffer and elute the adsorbed materials with 300ml of a linear gradient of 10-300mM phosphate. Collect 3.0- ml fractions of eluate. Note that the 26S proteasome can be adsorbed in the hydroxylapatite column under a low concentration of ATP and that the 20S proteasome and PA700 regulatory complex can be eluted separately at different phosphate concentrations of approximately 150 and 50mM, respectively. The 20S proteasome is detected by measuring the Suc-LLVYMCA- degrading activity with 0.05% SDS as described earlier, whereas the PA700 complex is monitored by immunoblotting with antibodies against their subunits. Collect protein in fractions containing the PA700 complex from hydroxylapatite chromatography, concentrate it to 2.0mg/ml by ultrafiltration with an Amicon PM-30 membrane, and subject samples of 1.0-2.0mg of protein to 10-30% glycerol density gradient centrifugation, similar to step 6 used in preparation of the 26S proteasome. Collect 1-ml fractions from the bottom of the centrifuge tube. The PA700 complex can be monitored by ATPase activity and/or the aforementioned immunoblotting analysis. Pool fractions 14-18 containing PA700 complex and store at -80°C.
Procedures
- Perfuse animal tissues with 25mM Tris-HCl buffer (pH 7.5) containing 1mM DTT, 1mM pheryhrethylsulforyl fluoride (PMSF), 20µg/ml E64, and 0.25 M sucrose and then homogenize in 3 volumes of the same buffer in a Potter-Elvehjem homogenizer. Centrifuge the homogenate for 1 h at 70,100g and use the resulting supernatant as the crude extract.
- Apply the crude extract directly to a Q-Sepharose column that has been equilibrated with buffer A. Wash the column with 5 bed volumes of buffer A and elute the adsorbed materials with a linear gradient of 0-0.8M NaCl in the same buffer. For detection of PA28, the activator of 20S proteasome, the hydrolysis of Suc- LLVY-MCA is assayed after preincubation for 10min at 4°C with approximately 0.5 µg of the latent 20S proteasome purified as described in Section IIIA. Eluate the PA28 activator with about 0.3 M NaCl. The endogenous activities of 20S and 26S proteasomes are monitored by assaying Suc-LLVY-MCA degradation with or without 0.05% SDS, respectively, which are coeluted at approximately 0.45 M NaCl.
- Combine the PA28 fractions from the QSepharose column, dialyze against buffer A, and apply it to a heparin-Sepharose CL-6B column that has been equilibrated in the same buffer. Recover PA28 in the flow-through fraction, as it does not bind to this resin.
- Apply the flow-through fraction from the heparin-Sepharose CL-6B column directly to the hydroxylapatite column that has been equilibrated with buffer B. Wash the column with 5 bed volumes of the same buffer and elute the adsorbed material with a linear gradient of 10-200mM phosphate. Pool fractions of PA28.
- Concentrate the PA28 activator from the hydroxylapatite column to 2.0mg/ml by ultrafiltration with an Amicon PM-10 membrane. Incubate the PA28 with the purified 20S proteasome (about 0.5 mg) for 30min at 4°C to form the PA28-20S proteasome complex and subject 2.0-mg samples of protein to 10-40% glycerol density gradient centrifugation in a manner similar to step 6 used in preparation of the 26S proteasome. Note that excess PA28 should be used for association with the 20S proteasome because the PA28-20S proteasome complex is hardly separated from the 20S proteasome, unlike the PA28 complex, by density gradient centrifugation analysis. Collect 1-ml fractions from the bottom of the centrifuge tube. The PA28-20S proteasome complex is monitored by assaying Suc-LLVYMCA degrading activity. Pool fractions 14-18 and store at -80°C.
Preparation of PA28
Apply the PA28-20S proteasome complex from the glycerol density gradient centrifugation, the final material for preparation of football proteasomes, directly to a Q-Sepharose column that has been equilibrated in buffer A and wash extensively with the same buffer. Elute the adsorbed material with a linear gradient of 0-0.8M NaCl in the same buffer because the 20S proteasome and PA28 are separated by this column operation. Eluate the PA28 activator with about 0.3 M NaCl (for details, see step 2). Pool fractions containing PA28 and store at -80°C.
Hybrid proteasomes can be reconstituted by purified PA28 (or recombinant PA2800 and 26S proteasomes. Incubate 26S proteasomes (single- and double-capped PA700-20S proteasome complexes) with PA28 or PA280α at 37°C for 15-30min in 20mM Tris-HCl (pH 7.5) buffer containing 2 mM ATP, 5 mM MgCl2, and 1 mM EDTA (Kopp et al., 2001; Cascio et al., 2001).
D. Preparation of PA200
Procedures
- Homogenize a 125-g sample of animal tissues in 2.4 volumes (300 ml) of 10 mM Tris-HCl buffer (pH 7.5) containing 1mM DTT, 0.25% Triton X-10G and three Complete protease inhibitor tablets in a Waring blender. Centrifuge the homogenate for 1 h at 100,000g and use the resulting supernatant as the crude extract.
- Apply the crude extract (400 ml) directly to TSKDEAE (1.6 × 50cm) that has been equilibrated with buffer E. Wash the column with buffer E and elute the material with a 1 liter linear gradient from 35 to 300 mM KCl in the same buffer. The proteasome activity is detected by measuring the Suc-LLVY-MCAdegrading activity. Monitor PA200 by immunoblotting with antibody PA200.
- Dilute pool fractions containing PA200 from the DEAE column by twofold with buffer F and apply to 5 ml of Q-Sepharose fast flow resin equilibrated with buffer E Elute bound proteins with 10ml of 0.5 M KCl in the same buffer.
- Apply the elution fractions from Q-Sepharose resin to a Superdex 200-pg (2.6 × 60cm) column that has been equilibrated with buffer E containing 250 mM KCl and 5% glycerol. Pool fractions containing PA200.
- Dilute fractions from the size column threefold with 5% glycerol in buffer F and apply to a Mono Q column (0.5 × 5cm). Wash the column with the same buffer and resolve in 12 column volumes at 30mM/ml from 35 to 200mM KCl and then reduce the gradient to 20mM/ml from 200-400mM KCl and increase to 30 mM/ml from 400-500 mM KCl.
- Subject samples of the Mono Q pools to 5-20% glycerol density gradient centrifugation (30ml in buffer E containing Complete protease inhibitor tablets). Centrifuge for 19 h at 85,500g in a SW28 rotor and collect 1-ml fractions from the bottom of the centrifuge tube. Monitor PA200 by measuring Suc-LLVYMCA degradation for incubation of indicated fractions with 200 ng of purified proteasome and/or an immunoblotting analysis. Pool fractions containing PA200 and store at -80°C.
Various fluorogenic peptides are suitable for the measurement of 20S proteasomal activity because proteasomes show broad substrate specificity. However, Suc-LLVY-MCA is recommended as a sensitive substrate. Latent 20S proteasomes can be activated in various ways (Coux et al., 1996). We recommend the use of SDS at low concentrations of 0.02-0.08% for the activation of Suc-LLVY-MCA breakdown; the optimal concentration depends on the enzyme source and the protein concentration used. The fluorogenic peptide (Suc-LLVY-MCA) can be used for assay of PA700-, PA28-, PA200-20S proteasome complexes, but these are active without any treatment, unlike latent 20S proteasomes. For a specific assay, ATP-dependent degradation of polyubiquitinated 125I-labeled lysozyme should be measured, although such an assay is not easy because three kinds of enzymes, E1 (Ub-activating), E2 (Ub-conjugating), and E3 (Ub-ligating) must be purified for the in vitro preparation of ubiquitinated substrate (for the procedures, see Tamura et al., 1991). There are various E2 and E3 enzymes. Most of them have not yet been characterized for use in in vitro-reconstituted proteolytic systems, so it is difficult to prepare large amounts of ubiquitinated proteins for use as substrates for 26S proteasomes. Therefore, for quantitative and sensitive measurement of ATP-dependent proteolysis activity in vitro in mammalian cells, ornithine decarboxylase (ODC) is a useful substrate. ODC is the only known natural substrate independent of ubiquitination for recognition and degradation by 26S proteasomes and hybrid proteasomes. Antizyme (AZ), an ODC inhibitory protein, however, is needed for the process instead of Ub, but both ODC and AZ, required for this in vitro degradation assay, are available as recombinant proteins (Murakami et al., 1999; Tanahashi et al., 2000). Note that AZ is not present in lower organisms, such as yeasts, and thus this assay is not fit for these cells. It is also possible that their purification is monitored by measuring ATPase activity at later steps of their purification, since the 26S proteasome and PA700 regulator complex have intrinsic ATPase activity.
For purification of the 26S proteasome, ATP (0.5 or 2mM), together with 20% glycerol and 1 mM DTT, should be added to all solutions used because they strongly stabilize the 26S proteasome complex; the purified enzyme is stable during storage at -70°C for at least 6 months in the presence of 2 mM ATP and 20% glycerol. Other drastic chromatographs should be avoided because these operations may result in dissociation of the 26S complex into its constituents. Alternative methods of purification of the PA28 and PA700 regulatory complexes have been reviewed by DeMartino and Slaughter (1993).
References
Baumeister, W., Walz, J., Zuhl, F., and Seemuler, E. (1998). The proteasome: Paradigm of a self-compartmentalizing protease. Cell 92, 367-380.
Bochtler, M., Ditzel, L., Groll, M., Hartmann, C., and Huber, R. (1999). The proteasome. Annu. Rev. Biophys. Biomol. Struct. 28, 295-317.
Cascio, P., Call, M., Petre, B. M., Walz, T., and Goldberg, A. L. (2002). Properties of the hybrid form of the 26S proteasome containing both 19S and PA28 complexes. EMBO J. 21, 2636-2645.
Coux, O., Tanaka, K., and Goldberg, A. L. (1996). Structure and functions of the 20S and 26S proteasomes. Annu. Rev. Biochem. 65, 801-847.
Finley, D., Tanaka, K., Mann, C., Feldmann, H., Hochstrasser, M., Vierstra, R., Johnston, S., Hampton, R., Haber, J., McCusker, J., Silver, P., Frontali, L., Thorsness, P., Varshavsky, A., Byers, B., Madura, K., Reed, S. I., Wolf, D., Jentsch, S., Sommer, T., Baumeister, W., Goldberg, A., Fried, V., Rubin, D. M., Glickman, M. H. and Toh-e, A. (1998). Unified nomenclature for subunits of the Saccharomyces cerevisiae proteasome regulatory particle. Trends Biochem. Sci. 23, 244-245.
Hershko, A., and Ciechanover, A. (1998). The ubiquitin system. Annu. Rev. Biochem. 67, 425-479.
Kloetzel, P. M. (2001). Antigen processing by the proteasome. Nature Rev. Mol. Cell. Biol. 2, 179-187.
Kopp, E, Dahlmann, B., and Kuehn, L. (2001). Reconstitution of hybrid proteasomes from purified PA700-20 S complexes and PA28αβ activator: Ultrastructure and peptidase activities. J. Mol. Biol. 13, 465-471.
Murakami, Y., Matsufuji, S., Hayashi, S., Tanahashi, N., and Tanaka, K. (1999). ATP-dependent sequestration of ornithine decarboxylase by the 26S proteasome, a process coupled to unfolding, is a prerequisite for the degradation. Mol. Cell. Biol. 19, 7216-7227.
Pickart, C. M. (2001). Mechanisms underlying ubiquitination. Annu. Rev. B iochem. 70, 503-533.
Rock, K. L., York, I. A., Saric, T., and Goldberg, A. L. (2002). Protein degradation and the generation of MHC class I-presented peptides. Adv. Immunol. 80, 1-70.
Tanahashi, N., Murakami, Y., Minami, Y., Shimbara, N., Hendil, K. B., and Tanaka, K. (2000). Hybrid proteasomes: Induction by interferon-7 and contribution to ATP-dependent proteolysis. J. Biol. Chem. 275, 14336-14345.
Tamura, T., Tanaka, K., Tanahashi, N., and Ichihara, A. (1991). Improved method for preparation of ubiquitin-ligated lysozyme as substrates of ATP-dependent proteolysis. FEBS Lett. 292, 154-158.
Tanaka, K. (1998). Molecular biology of the proteasome. Biochem. Biophys. Res. Commun. 247, 537-541.
Tanaka, K., and Kasahara, M. (1998). The MHC class I ligandgenerating system: Roles of immunoproteasomes and the interferon-γ-inducible proteasome activator PA28. Immunol. Rev. 163, 161-176.
Unno, M., Mizushima, T., Morimoto, Y., Tomisugi, Y., Tanaka, K., Yasuoka, N., and Tsukihara, T. (2002). The structure of the mammalian 20S proteasome at 2.75 A*P26 resolution. Structure 10, 609-618.
Ustrell, V., Hoffman, L., Pratt, G., and Rechsteiner, M. (2002). PA200, a nuclear proteasome activator involved in DNA repair. EMBO J. 21, 3516-3525.
Support our developers




