Isolation of Latex Bead- and Mycobacteria-Containing Phagosomes
Phagocytosis is a crucial mechanism by which specialized cells, especially macrophages, neutophils, and dendritic cells, remove ingested debris, dead cells, and pathogens from the body. Many pathogens are killed within the phagosome (phagolysosome) as a consequence of fusion between this organelle and lysosomes, which delivers hydrolases and the proton ATPase into the phagosome. An important technical advance has been the use of nondegradable latex beads as phagosome marker: these have facilitated studies both in cells and in vitro after phagosome isolation. This article describes how latex bead phagosomes are isolated for in vitro studies. It also summarizes recent approaches developed for the more difficult problem of isolating phagosomes containing live and killed mycobacteria.
II. BACKGROUND
In 1967 in Edward Korn's group a simple but crucial innovation for both phagosome analysis and the study of membrane organelles in general was introducted (Weisman and Korn, 1967a,b). They fed Acanthamoeba with latex beads and, following gentle homogenisation, could isolate a remarkably pure fraction of latex bead phagosomes (LBP) by a single centrifugation step using a discontinuous sucrose gradient. For efficient uptake of beads into (mostly) single bead phagosomes, the beads had to be between 0.13 and 0.26µm (smaller and larger beads were taken in more ineffectively) (summarized by Desjardins and Griffiths, 2003). Although this system was used intermittently in subsequent years, the method was not used extensively until reintroduced by Desjardins et al. (1994a,b).
A recent proteomic analysis by Desjardins and collages has revealed that LBP from J774 cells have about 1000 proteins, of which ≈600 have already been identified (Garin et al., 2001; Desjardins and Griffiths, 2003). They also contain at least 100 different lipids (Kuehnel et al., 2004; Anes et al., 2003).
Since 1994 a large number of papers have appeared in which LBP have been analyzed in different cells or have been used after isolation. By October 2003, Medline listed 73 papers that involved LBP; the actual number is certain to be significantly larger, as LBP are often used as controls for pathogen-containing phagosomes and are often not mentioned in the titles or abstracts to these papers.
This article summarizes the methods used to prepare different beads and to isolate the ensuing phagosomes. No other membrane organelle in the cell can be prepared with such ease and to such high purity after a single purification step. We subsequently discuss our recent efforts in isolating phagosomes enclosing mycobacteria, an approach first initiated by Chakraborty et al (1994).
A. Preparations of Latex Beads for Internalisation
Latex beads, either "naked" or coated with substances of interest, can be used for the generation of phagosomes. Our standard system uses avidin-coated latex beads. For coating beads with avidin, incubate 100µl of latex beads (10%, w/v) with 100µl 0.5M MES buffer, pH 6.8, 722µl of distilled water, and 50µl of avidin (10mg/ml in water) for 15min on a rotating wheel. Add 14µl of 10mg/ml N-(3-dimethylaminopropyl)- N-ethylcarbodiimide hydrochloride (EDAC) and allow incubation for another 1.5h. Subsequently, add another 14µl EDAC to the reaction and incubate for a further 1.5h. Stop the reaction by adding 1% Triton X-100 in 10mM Tris pH 9.4. Pellet the beads by centrifugation in a desktop centrifuge at full speed (13,000×g) and then resuspend in Triton/tris buffer. Subsequently, wash the beads three times in phosphate-buffered saline (PBS) (each time followed by a centrifugation step) and store at 4°C in PBS containing a nonspecific protein; we use 0.03% fish skin gelatin (Sigma G7765) and 0.02% sodium azide. If determination of protein content on the beads is necessary, an aliquot of beads can be taken for protein measurement before adding the storage solution.
For isolation of LBP, grow 20 × 20-cm petri dishes of macrophages (in our case J-774A.1 macrophages are used) in 50ml of culture medium supplemented with 10% fetal calf serum (FCS) and 2mM glutamine until the needed density of cells is reached. Use J774 macrophages at 50 × 106 cells per dish. For internalisation, dilute 1 ml of beads with 25ml of Dulbecco's Modified Eagle's Medium (DMEM) per dish. Uptake is routinely allowed for 1 h on a shaker at 5% CO2 at 37°C. Remove non internalised beads by intensive washing of the cells with PBS at 37°C. Thereafter, incubate cells with internalized beads until the desired phagosome age is reached in the presence of DMEM at 37°C and 5% CO2.
 |
| FIGURE 1 |
C. Cell Lysis and Latex Bead Phagosome Isolation
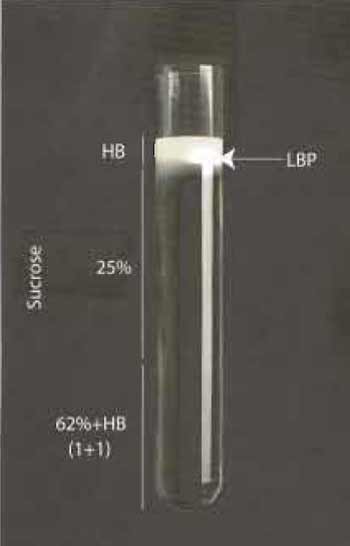
Harvest cells by adding 20ml PBS to the dish and scraping them with a suitable cell scraper. We use cell scrapers from Sarstedt (Nfimbrecht, Germany). Collect cells in 50-ml tubes and centrifuge at 1000×g for 10 min, resuspend in 5 ml of homogenization buffer (HB: 0.25 M sucrose, 3 mM imidazole, pH 7.4) with protease inhibitors (Table I) and pellet again, as before. Resuspend pellets in a final volume of 1.5 ml of HB buffer and lyse using 15-20 stokes with a 1-ml syringe fitted to a 22-gauge needle. Monitor cell breakage by light microscopy. Remove nuclei and intact cells by gentle centrifugation at 800×g at 4°C, collect supernatants and mix gently with 62% sucrose. Prepare the gradient in ultraclear centrifugation tubes from Beckmann (Reorder NO. 344060) as follows. Overlay 3-4ml of phagosome/62% sucrose mix with 5 ml of 25% sucrose containing protease inhibitors and overlay with 2 ml of HB buffer. Carry out centrifugation at 100,000g for 60 min at 4°C. The visible band that forms between the HB buffer and 25% sucrose contains the LBP and can be collected in approximately 200µl with a syringe by penetrating the tube wall with a 22-gauge needle (see Fig. 1). Aliquot phagosomes in 20 µl portions and store in liquid nitrogen until further use. For some purposes they can also be stored at -80°C; however, we have found that many activities are kept longer following liquid nitrogen storage.
 |
D. Monitoring Phagosomal Integrity
If it is necessary to check the proportion of intact phagosomes, phagosomes containing avidin-coated latex beads can be incubated conveniently with a suitable dye such as rhodamine-biotin 5nM for 15min at room temperature. The rationale here is that only broken phagosomes will be accessible to the label. Then the ratio of phagosomes labeled red (not intact) and nonlabelled phagosomes (intact) is determined by light microscopy. Routinely 70-80% of LBP are found to be intact in our experience.
1. Preparation of Mycobacteria for Uptake
There are a wide range of growth characteristics among the genus Mycobacterium, from 3h of generation time in M. smegmatis, 18-24h of M.tb, and 3 to 6 days for M. avium ssp. paratuberculosis or M. leprae, which has never been cultured in vitro. Usually, Middlebrook 7H10 (solid agar medium, Difco) or 7H9 (liquid medium, Difco) is enough for fast-growing species such as M. smegmatis or M. fortuitum, but for slow-growing species such as M. tuberculosis, M. avium, or BCG, enrich the medium with bovine serum albumin (BSA) fraction V and glucose prepared by dissolving 5g of albumin fraction V and 2 g glucose in distilled water that is brought to the final volume of 100ml. Sterilise this by passing it through a 0.45-µm Nucleopore filter and store in aliquots of 10ml at 4°C. When needed, for each 90ml of Middlebrook (7H9 or 7H10), use 10ml OADC (albumin, dextrose, catalase and oleic acid; Difco) to supplement the medium. For M. avium ssp. paratuberculosis, the medium has to be supplemented with 1 mg/liter Mycobactin J.
Grow bacterial cultures on petri dishes containing 4.7 g Middlebrook's 7H9 broth medium, 5 g of nutrient broth, supplemented with 0.5% glucose and 0.05% Tween 80 at 200rpm on a shaker at 37°C until the exponential grown phase (OD600 = 0.2) is reached (~108 cells / ml).
Subsequently, pellet and wash cells twice in PBS, pH 7.4, and resuspend in PBS to a final concentration of 5-10 × 109 cells/ml. Treat the suspension for 2 min in a water bath sonicator (room temperature) using four 30-s pulses to disperse clumps. Subsequently, bacteria should be passed through a 23-gauge needle to disrupt remaining bacterial clumps. Before infection, remove residual bacterial aggregates by lowspeed centrifugation (120g) for 2 min. The absence of bacterial clumps can finally be verified by light microscopy. It should be noted that some investigations have been interested in differences in the biology of mycobacteria that are taken up as aggregates rather than as individual bacteria (Schuller et al., 2001).
3. Preparation of a Suspension of Slow-Growing Mycobacteria such as M. tuberculosis
Inoculate 20 ml of Middlebrook's 7H9 broth medium with OADC with colonies isolated from Loewenstein Jennsen agar plates. In order to prevent the loss of lipidic virulence components, detergents such as Tween 80 should not be added the medium. Incubate the cultures at 37°C. When they reach the exponential phase growth (approximately 10-15 days) stock solutions in 10% glycerol can be preserved at -20°C up to 3 months or at -70°C for at least 1 year.
For some experiments it is useful to compare the biology of phagosomes containing live versus killed pathogens. Mycobacteria can be killed either by using paraformaldyde or, more easily, by heat. Suspensions resuspended in PBS (pH 7.4) are heat inactivated for 15min at 80°C. Such treatment completely blocks growth of bacteria as determined by colony-forming units, but keeps the appearance of the bacteria intact, as viewed by electron microscopy.
G. Labelling of Mycobaceria
For many experiments it is useful to fluorescently label bacteria before uptake. For this, wash freshly harvested bacteria twice (3000×g) in a Eppendorf centrifuge for 3 min in ice-cold PBS, pH 7,4 and resuspend in the same buffer. After harvesting, dilute mycobacteria using PBS to a OD600: 1.0. Add sulpho-NHS-FITC to a final concentration of 5µg/ml and allow labelling to continue for 20min on a shaker at room temperature. Non-incorporated FITC can be removed by washing in PBS plus 100 mM glycine. Thereafter, wash bacteria three times with PBS and immediately freeze in 100 µl aliquots at -20°C. To avoid batch variability in experiments, bacteria can be stored after labelling at -20°C up to 3 months or at -70°C for 1 year. The use of bacteria expressing GFP has emerged as a more powerful alternative to the surface labelling approach, as can be seen in numerous publications (see, e.g., Dhandayuthapani et al., 1995; Kremer et al., 1995).
The method we describe is a variation of that introduced by Chakraborty (1994). For preparing the mycobacteria-containing phagosome preparation, we usually infect macrophages of approximately 70% confluency in 245 × 245-mm dishes with mycobacteria. At desired times postinfection, wash cells in PBS. Harvest infected cells by scraping them in 20 ml of PBS and collecting them in one 50-ml tube per dish of cells and pellet by centrifugation at 800×g for 10min at 4°C. Resuspend cells in 5 ml of HB with protease inhibitors and pellet again. In order to have a good lysis of the infected macrophages in the next step, cells have to be resuspended in an optimal volume of HB. As a rule of thumb, use an equal volume of HB (with inhibitors) to resuspend the pellet of cells. This should result in approximately 1 ml of suspension. Variations of this ratio can be applied to optimize breakage of cells. Lyse macrophages by approximately 30 strokes with a syringe fitted with 22-gauge needle and check the percentage of broken cells by light microscopy (Fig. 1). The process should continue until roughly 80% of the cells are broken, but with intact nuclei (Fig. 2). Subsequently, dilute the suspensions with an equal volume of HB plus inhibitors and centrifuge at 300 g for 10 min at 4°C and discard the pellet containing nuclei and intact cells. The postnuclear supernatant (PNS) contains the phagosomes, as well as endocytic and other organelles and cytoplasmic components. It is important to note that loss of phagosomes at this stage occurs mainly because of two reasons. First, cells are not broken properly and phagosomes are not set free in the supernatant. This can be a result of a too dilute suspension, which is more resistant to breakage, or from too gentle application or not enough strokes with the syringe. Second, phagosomes can be lost due to breakage of nuclei. This results in free DNA in the suspension that binds to phagosomes and keeps the phagosomes in the pellet together with nuclei and intact cells. As a result, cell lysis is one of the most critical stages of isolating bacterial phagosomes from macrophages.
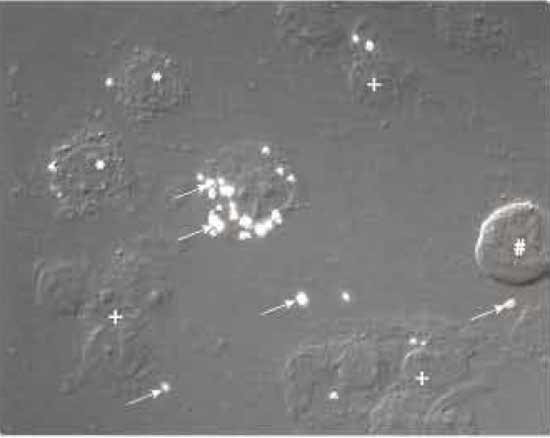 |
| FIGURE 2 Nomarski interference microscopy image of M. smegmatis-infected J774 macrophages treated with a syringe equipped with a 22-gauge needle 10 times. *, cells in the process of breaking; +, nuclei with associated cell debris; #, intact cell; arrows indicate M. smegmatis-containing phagosomes that are phase refractile. |
 |
| FIGURE 3 |
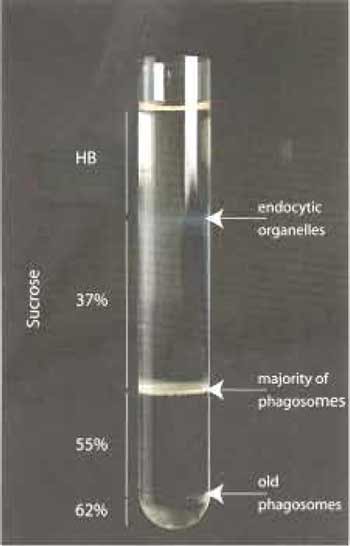
If a further concentration of the preparation is needed, a second step gradient can be done. The combined collected fractions (up to 2ml) can be layered on the top of 2 ml 55% sucrose and centrifuged again at 100,000×g for 30min at 4°C The band on the top of the 55% sucrose contains the phagosomes and is collected as described earlier. Phagosomes can be used directly or stored in aliquots in liquid nitrogen until further use.
The successful isolation of intact phagosomes also depends on minimizing the interaction of phagosomes with tube surfaces. One must avoid pelleting the phagosomes until the final stage in the isolation.
Latex bead phagosomes have emerged as a powerful model membrane organelle system that can be used to analyse many functions that are general properties of many intracellular membrane organelles, such as membrane fusion, their interaction with actin or microtubules, or acidification. The same particles can be used conveniently for in vitro analysis or for complementary studies in cells. In principle, any ligand that binds to cell surface receptors can be conjugated to beads in order to enrich for specific receptors in the phagosomes. It should be noted that the size of the particle is an important consideration; for phagocytosis, there is a minimum size (usually 0.2-0.3µm in diameter) as well as a maximum size that depends on the cell type (for cultures macrophages, this is usually around 5µm in diameter); for more details, see Desjardins and Griffiths (2003). A survey of the literature, as well as our own experience, convinces us that LBP are an excellent model system that behave similarly to phagosomes containing microorganisms that can be killed by macrophages (such as M. smegmatis; see Anes et al., 2003). LBP have also been used extensively as control phagosomes for analyses of pathogen-enclosing phagosomes; the latter usually modify the intracellular behaviour of the phagosome in a manner that facilitates the survival and growth of the pathogens. A key question that needs to be addressed individually for each intraphagosomal pathogen is how the pathogen manages to by-pass the normal phagosomal killing mechanisms. Also for this question the use of latex beads can be invaluable, as suspected pathogen surface "virulence" factors that bind to cell surface receptors can be conjugated to beads to see if and how the intracellular trafficking and molecular composition of the phagosome differ from control bead phagosomes.
References
A1-Haddad, A., Shonn, M. A., Redlich, B., Blocker, A., Burkhardt, J. K., Yu, H., Hammer, J. A., 3rd, Weiss, D. G., Steffen, W., Griffiths, G., and Kuznetsov, S. A. (2001). Myosin Va bound to phagosomes binds to F-actin and delays microtubule-dependent motility. Mol. Biol. Cell 12, 2742-2755.
Anes, E., Kuhnel, M. R, Bos, E., Moniz-Pereira, J., Habermann, A., and Griffiths, G. (2003). Selected lipids activate phagosome actin assembly and maturation resulting in killing of pathogenic mycobacteria. Nature Cell Biol. 5, 793-802.
Blocker, A., Griffiths, G., Olivo, J. C., Hyman, A. A., and Severin, F. F. (1998). A role for microtubule dynamics in phagosome movement. 1. Cell Sci. 111, 303-312.
Blocker, A., Severin, F. F., Burkhardt, J. K., Bingham, J. B., Yu, H., Olivo, J. C., Schroer, T. A., Hyman, A. A., and Griffiths, G. (1997). Molecular requirements for bi-directional movement of phagosomes along microtubules. J. Cell Biol. 137, 113-129.
Chakraborty, P., Sturgill-Koszycki, S., and Russell, D. G. (1994). Isolation and characterization of pathogen-containing phagosomes. Methods Cell Biol. 45, 261-276.
Defacque, H., Egeberg, M., Antzberger, A., Ansorge, W., Way, M., and Griffiths, G. (2000). Actin assembly induced by polylysine beads or purified phagosomes: Quantitation by a new flow cytometry assay. Cytometry 41, 46-54.
Desjardins, M., Celis, J. E., van Meer, G., Dieplinger, H., Jahraus, A., Griffiths, G., and Huber, L. A. (1994). Molecular characterization of phagosomes. J. Biol. Chem. 269, 32194-32200.
Desjardins, M., and Griffiths, G. (2003). Phagocytosis: Latex leads the way. Curr. Opin. Cell Biol. 15, 498-503.
Desjardins, M., Huber, L. A., Parton, R. G., and Griffiths, G. (1994). Biogenesis of phagolysosomes proceeds through a sequential series of interactions with the endocytic apparatus. J. Cell Biol. 124, 677-688.
Garin, J., Diez, R., Kieffer, S., Dermine, J. E, Duclos, S., Gagnon, E., Sadoul, R., Rondeau, C., and Desjardins, M. (2001). The phagosome proteome: Insight into phagosome functions. J. Cell Biol. 152, 165-180.
Greenberg, S., and Silverstein, S. C. (1993). Phagocytosis. In "Fundamental Immunology" Chap. 27, pp. 941-964.
Jahraus, A., Egeberg, M., Hinner, B., Habermann, A., Sackman, E., Pralle, A., Faulstich, H., Rybin, V., Defacque, H., and Griffiths, G. (2001). ATP-dependent membrane assembly of F-actin facilitates membrane fusion. Mol. Biol. Cell. 12, 155-170.
Jahraus, A., Tjelle, T. E., Berg, T., Habermann, A., Storrie, B., Ullrich, O., and Griffiths, G. (1998). In vitro fusion of phagosomes with different endocytic organelles from J774 macrophages. J. Biol. Chem. 273, 30379-30390.
Korn, E. D., and Weisman, R. A. (1967). Phagocytosis of latex beads by Acanthamoeba. II. Electron microscopic study of the initial events. J. Cell Biol. 34, 219-227.
Kremer, L., Baulard, A., Estaquier, J., Poulain-Godefroy, O., and Locht, C. (1995). Green fluorescent protein as a new expression marker in mycobacteria. Mol. Microbiol. 17, 913-922.
Schuller, S., Neefjes, J., Ottenhoff, T., Thole, J., and Young, D. (2001). Coronin is involved in uptake of Mycobacterium bovis BCG in human macrophages but not in phagosome maintenance. Cell Microbiol. 3, 785-793.
Weisman, R. A., and Korn, E. D. (1967). Phagocytosis of latex beads by Acanthamoeba. I. Biochemical properties. Biochemistry 6, 485-497.
Support our developers




