Isolation of Peroxisomes
The investigation of unique functional and structural aspects of peroxisomes (PO) requires the preparation of highly purified fractions of this organelle. This is, however, hampered by two serious problems: (1) the relative paucity of PO (2% of total liver protein) and (2) their considerable fragility. Thus, mild homogenization conditions minimizing mechanical, hydrostatic, and osmotic stress have to be sustained.
In general, the isolation of PO is accomplished in three steps: (a) homogenization of the tissue or disruption of the cells; (b) subfractionation of the homogenate by differential centrifugation usually according to the classical scheme of de Duve et al. (1955); and (c) isolation of purified peroxisomes by density gradient centrifugation of the so-called light mitochondrial (λ) fraction.
Homogenization is commonly carried out in an isotonic medium (e.g., see Section III,A,1) at low salt concentrations to avoid aggregation. Addition of a chelator (e.g., EDTA), however, is feasible, as it prevents the aggregation of microsomes that may contaminate PO. Moreover, the homogenization buffer (HB) should be supplemented with antioxidants (dithiothreitol, DTT), as well as protease inhibitors (phenylmethylsulfonyl fluoride, PMSF; ε-aminocaproic acid) to block oxidative and proteolytic activities in the course of the isolation procedure.
The method described in this article is a modification of the protocol established for the isolation of highly purified (>98%) PO from normal rat liver (Völkl and Fahimi, 1985). Meanwhile, it has been applied to livers and kidneys of several other mammalian species (Fahimi et al., 1993; Zaar et al., 1992), as well as for the isolation of PO from cell cultures (Schrader et al., 1994). With an additional differential centrifugation step, even peroxisomal subpopulations may be isolated according to this protocol, as has been demonstrated by Lüers et al. (1993).
A. Instrumentation
- Perfusion device (self-made)
- A 30-ml Potter-Elvehjem tissue grinder (Cat. No. 1931 05145) with a loose-fitting Teflon pestle (Cat. No. 1931 05155; clearance 0.10-0.15mm) and a motor-driven homogenizer (Cat. No. 5308 50001) are from Migge, 69123 Heidelberg, FRG.
- Refrigerated low- and high-speed centrifuge (e.g., Beckman TJ-6 and J 2-21); ultracentrifuge (e.g., Beckman L90) with corresponding rotors (e.g., Beckman JA-20; VTi 50).
- Refractometer
B. Chemicals
Morpholinopropane sulfonic acid (MOPS, Cat. No. 29 836), PMSF (Cat. No. 32 395), and DTT (Cat. No. 20 710) are from Serva (Heidelberg, FRG). Ethanol (Cat. No. 8006) and NaCl (Cat. No. 0277) are from J. T. Baker (Deventer, Netherlands). EDTA (Cat. No. E 2628-2) is from Max Keller (Mannheim, FRG), ε-aminocaproic acid (Cat. No. 62 075) is from Riedel-de Haen (Seelze, FRG), sucrose (Cat. No. 4621) is from Roth (Karlsruhe, FRG), and metrizamide (Cat. No. 10o 1983N) is from Life Technologies (76344 Eggenstein- Leopoldshafen).
C. Animals
Female rats of 220-250g body weight, starved overnight
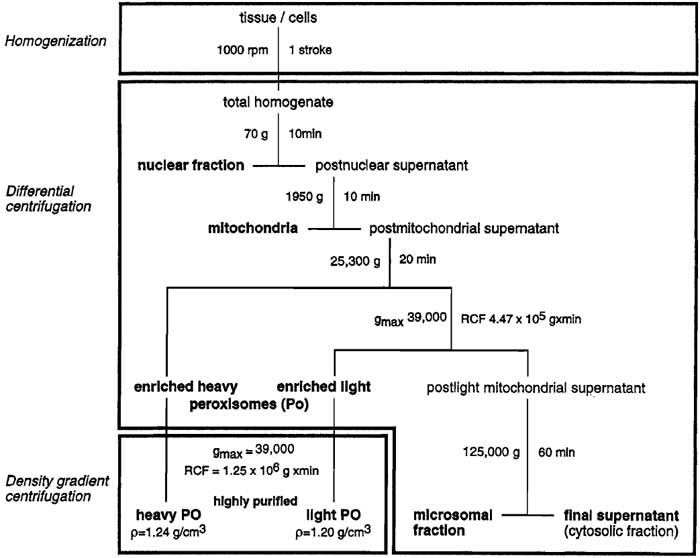 |
| FIGURE 1 Flowchart for the isolation of highly purified (>98%) peroxisomes from rat liver. |
A. Perfusion and Homogenization
Solutions
- Homogenization buffer (HB): To make 1 liter, dissolve 85.56g of sucrose (250mM), 1.046g of MOPS (5mM), 0.372 g of EDTA-Na2 (1mM), and 1 ml of ethanol (0.1%) in distilled water, adjust pH to 7.2 with NaOH, and add water up to 1 liter. Store at 4°C. Prior to use add per 100ml: 0.2µl of 0.1M PMSF, 0.1ml of 1M ε-aminocaproic acid, and 20µl of 1M DTT.
- Saline (0.9%): To make 1 liter, dissolve 9g of NaCl in distilled water and adjust to a total volume of 1 liter.
Steps
- Anesthesize the animal (e.g., by ip injection of chloralhydrate).
- Weigh the animal, open abdominal cavity, and perfuse liver with 0.9% saline via the portal vein until all blood is drained away.
- Remove liver, dissect connective tissue, and weigh and cut liver into small pieces collected in a Potter tissue grinder held in an ice bath containing 3 ml/g (wet liver weight) of ice-cold HB.
- Homogenize tissue with a loose-fitting pestle, applying a single down-and-up stroke at 1000rpm.
- Pour homogenate into a 50-ml centrifuge tube.
B. Subcellular Fractionation
Solution
HB as described in Section III,A.
- To remove debris, unbroken hepatocytes, and blood cells, as well as most of nuclei, centrifuge the total homogenate at 70g for 10min in a refrigerated low-speed centrifuge.
- Carefully pour off the supernatant (loose pellet !), resuspend pellet in 2ml/g of ice-cold HB, rehomogenize, and spin again under the same conditions.
- Pour off the second supernatant and combine it with the first one (postnuclear supernatant); discard the pellet.
- Centrifuge postnuclear supernatant at 1950g for 10min in a refrigerated high-speed centrifuge.
- Decant supernatant (firm pellet), resuspend pellet manually in 1 ml/g of ice-cold HB using a glass rod, and spin again at 1950g. The final pellet (heavy mitochondrial fraction) contains the majority of mitochondria, large microsomal sheets, and some remaining nuclei. The combined supernatants represent the postmitochondrial supernatant.
- Subject the latter to 25,300g for 20min; remove supernatant, including the reddish fluffy layer by suction; resuspend pellet in about 10 ml of ice-cold HB using a glass rod; and recentrifuge at 25,300g for 15min. Resuspend the final pellet in 5ml of ice-cold HB again by means of a glass rod, which comprises the enriched heavy peroxisomal = light mitochondrial (λ) fraction. The corresponding supernatant may be either used directly to prepare a microsomal fraction and a final supernatant (soluble proteins mostly of cytosolic origin) or processed further for the isolation of "light PO."
- To this extent it is centrifuged at an integrated relative centrifugal force (RCF) of 4.47 × 105gmin (gmax = 39,000). Resuspend the pellet thus obtained in 5 ml of ice-cold HB using a glass rod; this pellet represents the enriched light peroxisomal fraction (Luers et al., 1993).
Solutions
- Gradient buffer (GB): To make 1 liter, dissolve 1.046g of MOPS (5 mM), 0.372g of EDTA-Na2 (1 mM), and 1 ml of ethanol (0.1%) in distilled H2O, adjust with NaOH to pH 7.2, and add H2O up to 1 liter. Store at 4°C. Prior to use add per 100ml: 0.2ml of 0.1M PMSF, 0.1 ml of 1M ε-aminocaproic acid, and 20 µl of 1M DTT.
- Metrizamide solutions (MS)
- 60% (w/v) stock solution: Dissolve 60g of metrizamide in GB by stirring. Add GB up to 100ml. Store at 4~
- Gradient solutions: To prepare one gradient, take 3.78, 3.38, 3.53, 2.06, and 3.2 ml of the 60% stock solution. Add up GB to 10, 7, 6, 3, and 4ml using a refractometer to definitely adjust densities to 1.12, 1.155, 1.19, 1.225, and 1.26 g/ml, respectively.
Steps
Preparation of a Metrizamide Gradient
- Layer sequentially 4, 3, 6, 7, and 10ml, respectively, of MS (1.26-1.12g/ml) in a 40-ml centrifuge tube (e.g., Quick-seal polyallomer, Beckman) to form a discontiuous gradient.
- Immediately freeze the gradient in liquid nitrogen and store it at -20°C.
- Thaw the gradient quickly at room temperature using a metallic stand, thus transforming the step gradient into one with an exponential profile.
- Layer 5ml of either the light or the heavy enriched peroxisomal fraction (corresponding to one liver of approximately 5-6 g) on top of the thawed gradient and seal the tube.
- Centrifuge gradients in a vertical-type rotor (e.g., Beckman VTi50) at an integrated force of 1.256 × 106g min (gmax= 39,000) using slow acceleration/deceleration modes. Under the conditions employed, highly purified heavy peroxisomes band at 1.23-1.24g/ml and light peroxisomes band at 1.20-1.21 g/ml.
- Recover the peroxisomal fraction by means of a fraction collector; alternatively, the gradient tube can be punctured and the fraction aspirated by a syringe. Store fraction at -80°C.
- To remove metrizamide, which interferes with the determination of some peroxisomal enzymes (e.g., urate oxidase) or of protein (Lowry method), dilute the peroxisome fraction about 10-fold with HB followed by centrifugation at 25,000 and 39,000 g, respectively, to pellet the organelles.
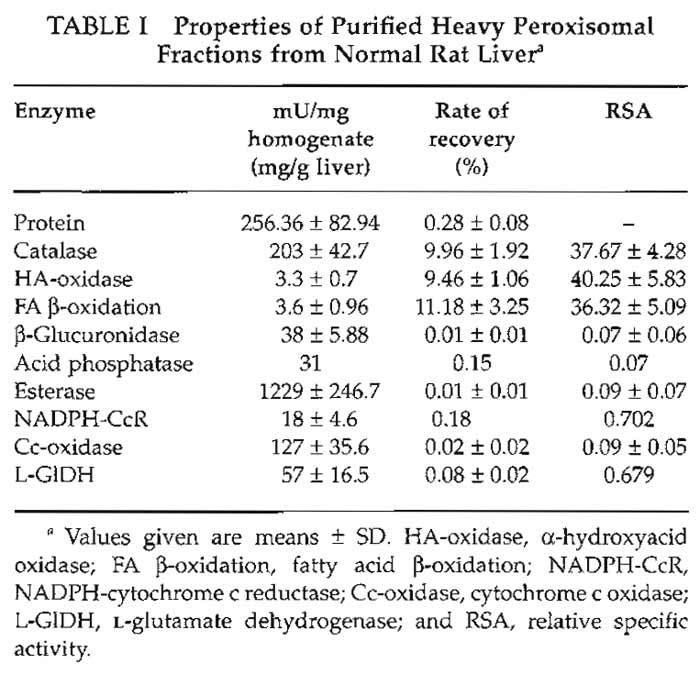
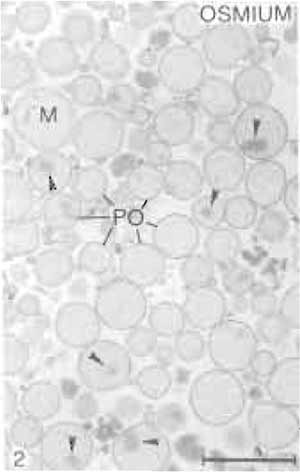
The properties of the heavy peroxisomal fraction are listed in Table I. Estimated by the specific peroxisomal reference enzymes, it shows a purification rate of about 38-fold over the original homogenate. More than 95% of the total protein content of this fraction is contributed by peroxisomes (Völkl and Fahimi, 1985), with mitochondria and microsomes accounting for about 2% each and lysosomes for less than 1%. This is confirmed by electron microscopy, which shows that peroxisomes make up 98-99% of the fraction (Figs. 2 and 3). Many peroxisomes contain the typical inclusions of urate oxidase in the matrix (Fig. 2), but some extruded free cores are also found between the organelles. The electron-dense cytochemical reaction product of catalase after the incubation of filter preparations in the alkaline 3,3'-diaminobenzidine medium (Fahimi, 1969) is seen over the matrix of the majority of peroxisomes (Fig. 3), demonstrating their integrity and the absence of leakage of catalase.
 |
|
|
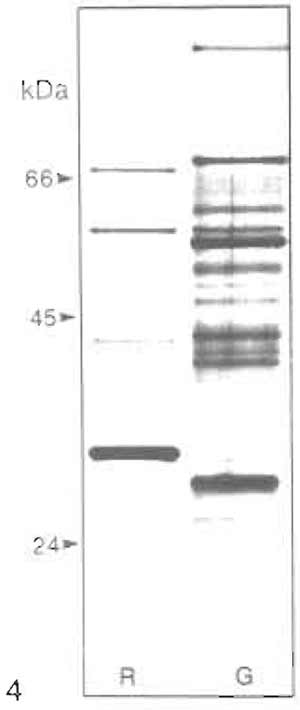
The polypeptide pattern (SDS-PAGE) of rat liver heavy peroxisomes is shown in Fig. 4 (lane R) confirming their high degree of purity because of the absence of bands typical for mitochondria and microsomes. It also shows distinct differences in protein composition of hepatic peroxisomes between rat (R) and guinea pig (Fig. 4, lane G). The selective induction of specific peroxisomal proteins, such as the trifunctional protein (PH) in rats treated with hypolipidemic fibrates (lane Bz) with the concomitant reduction of catalase (Cat) and urate oxidase (UOx), is apparent in Fig. 5.
|
|

Acknowledgments
The original work in the laboratory of the authors has been supported by grants of the Deutsche Forschungsgemeinschaft, Bonn, FRG (Fa 146/1-3; Vo 317/3-1; SFB 352 and Vo 317/4-1) and Landesforschungsschwerpunkt- Programm of the State of Baden-Württemberg, FRG.
References
De Duve, C., Pressman, B. C., Gianetto, R., Wattiaux, R., and Appelmans, F. (1955). Intracellular distribution patterns of enzymes in rat liver tissue. Biochem. J. 60, 604-617.
Fahimi, H. D. (1969). Cytochemical localization of peroxidatic activity of catalase in rat hepatic microbodies (peroxisomes). J. Cell Biol. 43, 275-288.
Fahimi, H. D., Baumgart, E., Beier, K., Pill, J., Hartig, F., and Völkl, A. (1993). Ultrastructural and biochemical aspects of peroxisome proliferation and biogenesis in different mammalian species. In "Peroxisomes: Biology and Importance in Toxicology and Medicine" (G. G. Gibson and B. Lake, eds.), pp. 395-424. Taylor and Francis, London.
Hartl, F. U., Just, W. W., Köster, A., and Schimassek, H. (1985). Improved isolation and purification of rat liver peroxisomes by combined rate zonal and equilibrium density centrifugation. Arch. Biochem. Biophys. 237, 124-134.
Leighton, F., Poole, B., Beaufay, H., Baudhuin, P., Coffey, J. W., Fowler, S., and De Duve, C (1968). The large-scale preparation of peroxisomes, mitochondria and lysosomes from the livers of rats injected with Triton WR-1339. J. Cell Biol. 37, 482-513.
Lüers, G., Hashimoto, T., Fahimi, H. D., and Völkl, A. (1993). Biogenesis of peroxisomes : Isolation and characterization of two distinct peroxisomal populations from normal and regenerating rat liver. J. Cell Biol. 121, 1271-1280.
Schrader, M., Baumgart, E., Völkl, A., and Fahimi, H. D. (1994). Heterogeneity of peroxisomes in human hepatoblastoma cell line HepG2: Evidence of distinct subpopulations. Eur. Cell Biol. 64, 281-294.
Titorenko, V., Ogrydziak, D. M., and Rachubinski, R. A. (1997). Four distinct secretory pathways serve protein secretion, cell surface growth, and peroxisome biogenesis in the yeast Yarrowia lipolytica. Mol. Cell Biol. 17, 5210-5226.
Van Veldhoven, P., Debeer, L. J., and Mannaerts, G. P. (1983). Waterand solute- accessible spaces of purified peroxisomes. Biochem. J. 210, 685-693.
Van Veldhoven, P., Baumgart, E., and Mannaerts, G. P. (1996). Iodixanol (Optiprep), an improved density gradient medium for the isoosmotic isolation of rat liver peroxisomes. Anal. Biochem. 237, 17-23.
Völkl, A., and Fahimi, H. D. (1985). Isolation and characterization of peroxisomes from the liver of normal untreated rats. Eur. J. Biochem. 149, 257-265.
Völkl, A., Mohr, H., Weber, G., and Fahimi, H. D. (1997). Isolation of rat hepatic peroxisomes by means of free flow electrophoresis. Electrophoresis 18, 774-780.
Wanders, R. J. A., Schutgens, R. B. H., and Barth, P. G. (1995). Peroxisomal disorders: A review. J. Neuropathol. Exp. Neurol. 54, 726-739.
Zaar, K. (1992). Structure and function of peroxisomes in the mammalian kidney. Eur. J. Cell Biol. 59, 233-254.
Support our developers




