Isolation and Subfractionation of Plasma Membranes to Purify Caveolae Separately from Lipid Rafts
Cellular membranes contain distinct microdomains with unique molecular topographies, including caveolae and lipid rafts (Fig. 1). Caveolae are specialized flask-shaped invaginated microdomains located on the cell surface membrane of many cell types that appear to form through the polymerization of caveolin (Monier et al., 1995), have a dynamin collar around their necks for fission (Oh et al., 1998), and contain other molecular machinery for vesicular budding, docking, and fusion, including VAMP-2, NSF, and SNAP (Schnitzer et al., 1995a). In contrast, lipid rafts are flat domains without the characteristic omegashaped form of caveolae that suggests possible trafficking function. Unlike caveolae, which form via protein-protein interactions, lipid rafts appear to form in the membrane when highly saturated sphingolipids self-assemble with cholesterol to pack into a highly ordered lipid phase (Brown and London, 2000). Caveolae and lipid rafts may share similar lipids but a detailed compositional analysis is lacking at this time.
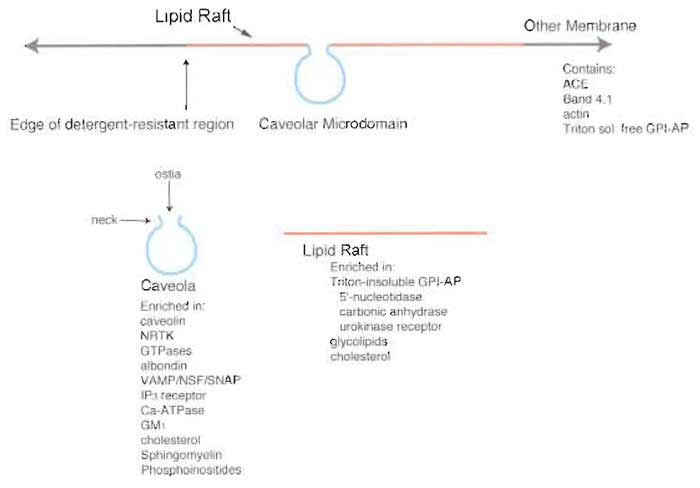 |
| FIGURE 1 Microdomain structure of the plasma membrane. The plasma membrane contains various low-density microdomains, predominantly caveolae and lipid rafts rich in GPI-AP, that exist as distinct regions of the cell surface. Although caveolae and lipid rafts exhibit similar buoyant densities and detergent resistance, they each contain a distinct protein expression profile that differs from each other as well as from the rest of the plasma membrane proper. NRTK, nonreceptor tyrosine kinases; ACE, angiotensin-converting enzyme; VAMP, vesicle-associated membrane protein; NSF, N-ethylmaleimide-sensitive fusion protein; SNAP, soluble NSF attachment protein; IP3, inositol 1,4,5-trisphosphate. |
II. MATERIALS AND INSTRUMENTATION
A. Materials
Sodium chloride (Cat. No. BP358), potassium chloride (Cat. No. BP366), magnesium sulfate (Cat. No. BP213), glucose (Cat. No. BP350), EDTA disodium salt (Cat. No. BP120), sodium bicarbonate (Cat. No. BP328), sucrose (Cat. No. BP220), magnesium chloride (Cat. No. BP214), Tris (Cat. No. BP152), methanol (Cat. No. A412), hydrochloric acid (Cat. No. A144), sodium hydroxide (Cat. No. SS255), sodium carbonate (Cat. No. BP357), 2-mercaptoethanol (Cat. No. BP176), Tricine (Cat. No. BP315), Tween 20 (Cat. No. BP337), silver nitrate (Cat. No. $181), glacial acetic acid (Cat. No. A38), sodium phosphate dibasic (Cat. No. BP332), potassium phosphate monobasic (Cat. No. BP362), potassium phosphate dibasic (Cat. No. BP363), Tygon tubing (Cat. No. 14-169-1B), razor blades (Cat. No. 12- 640), and 15-ml centrifuge tubes (Cat. No. 05-539-5) are from Fisher. HEPES (Cat. No. 737 151) and Pefabloc SC (Cat. No. 1 585 916) are from Roche Applied Science. 2-(N-Morpholine)ethanesulfonic acid (MES) (Cat. No. M2933), leupeptin (Cat. No. L 2884), O-phenanthroline (Cat. No. P9375), pepstatin A (Cat. No. P 4265), sodium nitroprusside (Cat. No. S 0501), E-64 (trans-epoxysuccinyl- L-leucylamido-[4-guanidino] butane) (Cat. No. E 3132), glycerol (Cat. No. G 5516), potassium acetate (Cat. No. p 3542), magnesium acetate (Cat. No. M 0631), and EGTA (Cat. No. E 4378) and from Sigma. Percoll (Cat. No. 17-0891-01) is from Amersham Biosciences. Protease inhibitor Cocktail I (2004) is from EMD Biosciences (Cat. No. 539131) Ten percent Triton X-100 (Cat. No. 28314) is from Pierce. Goat antimouse IgG-HRP conjugate (Cat. No. NA931) and goat antirabbit IgG-HRP conjugate (Cat. No. NA934) are from Amersham. Goat antimouse IgG-bodipy conjugate (Cat. No. B-2752), goat antimouse IgG-Texas red conjugate (Cat. No. T-862), goat antirabbit IgG-bodipy conjugate (Cat. No. B-2766), and goat antirabbit IgG-Texas red conjugate (Cat. No. T-2767) are from Molecular Probes. Polyacrylic acid (PAA) (Cat. No. 00627) is from Polysciences Inc. Nycodenz (Cat. No. AN-7050/BLK) is from Accurate Chemicals. Sprague Dawley rats (male) are from Charles Riven SW28 tubes (Cat. No. 344058) and SW55 tubes (Cat. No. 344057) are from Beckman. Ketamine is from Fort Dodge Animal Health and xylazine is from Vedco Inc. Nitex filters 53 µm (Cat. No. 3-50/31) and 30µm (Cat. No. 3-30/18) are from Sefar America Inc. Type "AA" homogenizer (Cat. No. 3431-E10), type "BB" homogenizer (Cat. No. 3431- E20), and type "C" homogenizer (Cat. No. 3431-E25) are from Thomas Scientific. Silk suture (3-0) is from Ethicon. Aluminum tubing (Cat. No. 89-78K101) is from McMaster-Carr. The three-way stopcock (Cat. No. 732-8103) is from Bio-Rad.
Ultracentrifugation is carried out in a Beckman L8- 80, Sorvall Pro 80, or Optima MaxE ultracentrifuge. Other centrifugation is carried out in a Brinkman Eppendorf 5415C or IEC Centra MP4R.
C. Perfusion Apparatus
On a ring stand, attach five 60-cc syringes attached to a series of five three-way stopcocks by way of Tygon tubing (0.0625in. i.d.). The series of three-way stopcocks are attached to Tygon tubing with an stainless steel loop (0.046 in. i.d.) inserted in the middle of the tubing. The stainless-steel loop is used to regulate the temperature of the solutions perfused into the rat vasculature by emersion in temperature-controlled water. The pressure of flow is measured by a sphygmomanometer attached to the 60-cc syringes by Tygon tubing and a single hole rubber stopper. The flow for perfusion is normally 8 mm of Hg. The flow rate at 25°C is approximately 5 ml/min and at 10°C is 4 ml/min.
Unless otherwise specified in the protocol, all procedures should be performed at 4°C and all solutions must be kept and used ice cold.
Solutions
Buffer PM: 0.25 M sucrose, 1 mM EDTA, 20 mM Tricine in ddH2O pH to 7.8 with NaOH (filter through 0.45-µm bottle top filter (can be stored at 4°C). For 500ml: 42.79g sucrose, 0.186g EDTA (disodium salt), and 1.79 g Tricine.
Colloidal silica solution: From a 30% positively charged colloidal silica stock solution, dilute to 1% with MBS pH 6.0 [pH to 6.0 with NaOH if necessary and filter through 0.45-µm bottle top filter (can be stored at 4°C). For 300ml: 10ml stock silica in 490 ml MBS, pH 6.0, with pH strips (usually you do not have to adjust pH).
Cytosolic buffer: 25mM potassium chloride, 2.5mM magnesium acetate, 5mM EGTA, 150mM potassium acetate, 25mM HEPES in ddH2O pH to 7.4 with KOH and filter through 0.45-µm bottle top filter (can be stored at 4°C). For 500ml: 0.93g KCl, 0.27 g Mg(C2H3O2)2·4H2O, 0.95 g C14H24N2O10, 7.36 g KC2H2O2, and 2.98 g HEPES.
250mM HEPES, pH 7.4: 29.8g in 450ml ddH2O, pH to 7.4 with NaOH, bring to 500 ml with ddH2O
1M KCl: 7.46 g in 100 ml ddH2O
Mammalian Ringer's solution (without calcium): 114mM sodium chloride, 4.5mM potassium chloride, 1 mM magnesium sulfate, 11mM glucose, 1.0 mM sodium phosphate (dibasic), 25 mM sodium bicarbonate in ddH2O pH to 7.4 with HCl (filter through 0.45-µm bottle top filter (can be stored at 4°C). For 2 liters: 13.32g NaCl, 0.67g KCl, 0.492g MgSO4·7H2O, 3.96g glucose, 0.28g Na2HPO4, and 4.20 g NaHCO3.
MES buffered saline (MBS): 20 mM MES, 135 mM NaCl in ddH2O pH to 6.0 with NaOH [filter through 0.45- µm bottle top filter (can be stored at 4°C). For 2 liters: 7.8 g MES and 14.6 g NaCl.
80% Nycodenz: 8ml 100% Nycodenz, 200µl 1M KCl bring to 10 ml with ddH2O
75% Nycodenz: 7.5ml 100% Nycodenz, 200µl 1M KCl bring to 10 ml with ddH2O
70% Nycodenz: 7ml 100% Nycodenz, 200µl 1M KCl bring to 10 ml with ddH2O
60% Nycodenz: 6ml 100% Nycodenz, 200µl 1M KCl bring to 10 ml with ddH2O
100% Nycodenz (v/w): 100g Nycodenz in 100ml of ddH2O
70% Nycodenz/sucrose/HEPES: 7ml 100% Nycodenz, 1 ml 60% sucrose, 1 ml 250mM HEPES, pH 7.4, 200 µl 1M KCl bring to 10 ml with ddH2O
65% Nycodenz/sucrose/HEPES: 6.5ml 100% Nycodenz, 1 ml 60% sucrose, 1 ml 250mM HEPES, pH 7.4, 200 µl 1M KCl bring to 10ml with ddH2O
60% Nycodenz/sucrose/HEPES: 6ml 100% Nycodenz, 1 ml 60% sucrose, 1 ml 250mM HEPES, pH 7.4, 200 µl 1M KCl bring to 10 ml with ddH2O
55% Nycodenz/sucrose/HEPES: 5.5ml 100% Nycodenz, 1 ml 60% sucrose, 1 ml 250mM HEPES, pH 7.4, 200 µl 1M KCl bring to 10ml with ddH2O
Phosphate-buffered saline (PBS): 137mM sodium chloride, 2.7mM potassium chloride, 4.3 mM sodium phosphate (dibasic), 1.4mM potassium phosphate (monobasic) in ddH2O pH to 7.4. For 1 liter: 8 g sodium chloride, 0.2 g potassium chloride, 0.61 g sodium phosphate (dibasic), 0.2g potassium phosphate (monobasic).
Polyacrylic acid: From 25% stock solution, dilute to 0.1% with MBS, pH 6.0 [pH to 6.0 with NaOH and filter through 0.45-µm bottle top filter (can be stored at 4°C)]. For 500ml: 2ml stock 25% polyacrylic acid in 480ml MBS, pH 6.0, pH to 6.0 and bring to 500ml with MBS pH 6.0.
Potassium phosphate/polyacrylic acid: 4 M potassium phosphate (dibasic) 2% PAA in ddH2O pH to 11 with NaOH. For 10 ml: 6.96 g potassium phosphate (dibasic) and 0.8ml 25% stock polyacrylic acid.
60% sucrose (w/w): 385.95 g sucrose in 200ml ddH2O, add 10ml 1M KCl, after the sucrose has dissolve, bring to 50ml with ddH2O
60% sucrose (w/w)/20mM KCl: 385.95g sucrose in 200ml ddH2O, add 10ml 1M KCl, after the sucrose has dissolve, bring to 50ml with ddH2O
35% sucrose (w/w)/20mM KCl: 5.2ml 60% sucrose, 200 µl 1M KCl, bring to 10ml with ddH2O
30% sucrose (w/w)/20mM KCl: 4.4ml 60% sucrose, 200µl 1M KCl, bring to 10ml with ddH2O
25% sucrose (w/w)/20mM KCl: 3.8ml 60% sucrose, 200µl 1M KCl, bring to 10ml with ddH2O
20% sucrose (w/w)/20mM KCl: 2.8ml 60% sucrose, 200µl 1M KCl, bring to 10ml with ddH2O
15% sucrose (w/w)/20mM KCl: 2.1ml 60% sucrose, 200µl 1M KCl, bring to 10ml with ddH2O
10% sucrose (w/w)/20mM KCl: 1.3ml 60% sucrose, 200µl 1M KCl, bring to 10ml with ddH2O
5% sucrose (w/w)/20mM KCl: 0.7ml 60% sucrose, 200µl 1M KCl, bring to 10ml with ddH2O
A. Purification of Silica-Coated Plasma Membranes (P)
1. Perfusion Procedure
- Anesthetize rats with a cocktail of ketamine (60mg/kg) and xylazine (1.6mg/kg). Perform a tracheotomy to ventilate the lungs using a respirator.
- Insert tubing from the perfusion apparatus into the pulmonary artery via the right ventricle and fasten by tying a 3-0 silk suture around the artery. Cut the left atrium to allow the flow to exit.
- Perfuse (4 ml/min) with Ringer's for 5 min starting at room temperature. After 1-1.5 min, begin lowering the temperature to ~10°C by placing the stainless- steel loop into an ice-cold bath. Gently drip ice-cold PBS over the lung to prevent drying. The remainder of the procedure should be performed at this cool temperature.
- Perfuse MBS for 1.5 min.
- Perfuse 1% colloidal silica solution for 1.5 min. At this point, keep the lungs inflated by replacing the tube attached to the respirator with a syringe and inflate with 3-5 ml of air.
- Flush with MBS for 1.5 min.
- Perfuse 0.1% PAA for 1.5 min.
- Flush lungs with 8ml of sucrose/HEPES with protease inhibitor Cocktail I (1×).
- Excise lungs from the animal and keep cold by immersion in ice-cold sucrose/HEPES with protease inhibitor Cocktail I (1×).
- Finely mince the excised lung mince with a "new" razor blade on a cold aluminum block embedded in ice.
- Add 20ml of sucrose/HEPES with protease inhibitors and place into a type "C" homogenizer vessel.
- Homogenize for 12 strokes at 1800rpm in a cold (4°C) room.
3. Purification of Silica-Coated Luminal Endothelial Cell Plasma Membrane (P)
- Filter the homogenate through a 53-µm Nytex filter, followed by a 30-µm Nytex filter.
- Remove 200µl from the filter solution, label "homogenate," and store at -20°C. Adjust the volume of the remaining solution to 20 ml with cold sucrose/HEPES with protease inhibitors.
- Add an equal volume of 100% Nycodenz and mix (this is enough for two SW28 tubes). Layer onto a 70-55% continuous Nycodenz sucrose/HEPES gradient (form by placing 3ml of 70, 65, 60, and 55 Nycodenz sucrose/HEPES and carefully swirling the solution, holding the tube at a 45° angle about 5-10 times).
- Top with sucrose/HEPES with protease inhibitors. Spin at 15,000rpm for 30min at 4°C in a SW28 rotor. Aspirate off the supernatant. Resuspend the pellet in 1 ml MBS. Add equal volume of 100% Nycodenz and mix.
- Layer onto a 80-60% continuous Nycodenz gradient (form by placing 350 µl of 80, 75, 70, 65, and 60% Nycodenz and twirling tube about 5-10 times). Top with 20mM KCl. Spin at 30,000rpm for 30min at 4°C in a SW55 rotor. Aspirate and discard the supernatant. Resuspend the pellet in 1 ml MBS and label as "P." Store at -20°C.
This protocol is written for T75 flasks; alter proportionately for larger or smaller flasks. All parts of the procedure need to be performed on ice or in a 4°C room.
- Wash cell monolayer three times with MBS.
- Overlay with 1% ice-cold colloidal silica solution and incubate for 10min. Wash cell monolayer three times with MBS.
- Overlay with 0.1% PAA and incubate for 10 min. Wash cell monolayer three times with MBS.
- Add 5ml of sucrose/HEPES with protease inhibitors (1×). Scrape cells and place in 15-ml centrifuge tube. Centrifuge 1000g for 5 min at 4°C.
- Bring to 1 ml with cold sucrose/HEPES with protease inhibitors (1×) (you can use up to six T75 flasks per 1 ml of cold sucrose/HEPES with protease inhibitors).
- Place in type "AA" homogenizer vessel. Homogenize with corresponding grinder for 20 strokes at 1800rpm.
- Remove 100 µl and label as "homogenate" and store at -20°C.
- Add an equal volume of 100% Nycodenz and mix (SW55 tube). Layer onto a 70-55% continuous Nycodenz sucrose/HEPES gradient (form by placing 350µl of 70, 65, 60, and 55% Nycodenz sucrose/HEPES and carefully twirling tube as described earlier).
- Top with sucrose/HEPES with protease inhibitors. Spin at 30,000rpm for 30 min at 4°C in a SW55 rotor. Aspirate off the supernatant. Resuspend the pellet in 1 ml MBS and label as "P." Store P at -20°C.
1. Isolation of Caveolae (V) (in the Presence of Detergent)
- Take 900 µl "P" (as per step 5 of Section III,A,3 or step 6 of Section III,A,4), add 100µl 10% Triton X-100, and mix. Save 100 µl "P." Mix on nutator for 10 min at 4°C. Place in type "AA" homogenizer vessel. Homogenize with corresponding grinder for 20 strokes at 1800 rpm.
- Bring to 40% sucrose (w/w) with 60% sucrose (w/w)/20mM KCl (takes about 1.5ml). Layer with discontinuous 35-0% sucrose (w/w)/20 mM KCl gradient (350µl of 35, 30, 25, 20, 15, 10, and 5 % sucrose / 20 mM KCl and 20 mM KCl).
- Spin at 50,000rpm for 4h at 4°C in a MLS-50 rotor.
- Collect the band between 10 and 15% sucrose (w/w) density. Dilute the band material two to three times with MBS and spin at 15,000rpm for 2h at 4°C. Resuspend pellet in 100µl MBS, pH 6.0, and label as "V"
- Collect and resuspend membrane pellet from step 3 in 1 ml MBS and label as "P-V."
- Store both fractions at -20°C.
2. Isolation of V" (Detergent Free)
- Process as in Section III,B,1 except omit the addition of Triton X-100 and homogenize for 60 strokes. Label floated caveolae as "V'" (V prime).
- Collect and resuspend membrane pellet from step 3 in Section III,A,4 and label as "P-V.'"
- Store V' and P-V' at -20°C.
1. Isolation of Rat Lung Cytosol
- Perfuse rat lung as described in Section III,A using steps 1, 2, 3, 8, and 9 only.
- Homogenize lung in cytosolic buffer (2-3×, v/w) for 10 strokes at 1800 rpm.
- Spin at 35,000rpm for 1 h at 4°C in a SW55 rotor. Place supernatant over PD-10 column and collect first peak. Aliquot (1 ml), label at "rat lung cytosol," and store at -80°C.
2. Isolation of "Vb"
- Incubate "P" or "PM" (PM described later) (20- 50µg/ml) with rat lung cytosol (1-5mg/ml) for 1 h at 37°C.
- Bring to 40% sucrose (w/w) with 60% sucrose (w/w)/20mM KCl solution. Layer with discontinuous 35-0% sucrose 20mM KCl gradient (as described in Section III,B; 350µl each layer).
- Spin at 30,000rpm overnight at 4°C in a SW55 rotor. Collect the band between 10 and 15% sucrose (w/w) density. Dilute the band material two to three times with MBS and spin at 15,000rpm for 2 h at 4°C. Resuspend pellet in 100µl MBS and label as "Vb."
- Collect and resuspend membrane pellet from step 2 in 1 ml MBS and label as "P-Vb." Store at -20°C.
Isolation of LR
- Resuspend membrane pellet (P-V) from Section III,B in 1 ml of ice-cold MBS, pH 6.0. Add equal volume of potassium phosphate/polyacrylic acid (pH should be greater than 9.5). Mix well.
- Sonicate at maximum output for 10 bursts of 10s each. Cool on ice between bursts. Mix using nutator at room temperature for 8h and then sonicate as described earlier except use 5- to 10-s bursts.
- Add Triton X-100 to a final concentration of 1%. Mix on nutator for 10 min at 4°C and then place in a type "AA" homogenizer vessel.
- Homogenize with corresponding grinder for 30 strokes at 1800rpm. Bring to 40% sucrose (w/w) using 60% sucrose (w/w)/20 mM KCl and layer with discontinuous 35-0% sucrose (w/w) with 20mM KCl using 350µl solution per layer as described Section III,A.
- Centrifuge at 30,000 rpm overnight at 4°C in a SW55 rotor.
- Collect band between 10 and 15% sucrose (w/w) density. Dilute the band material two to three times with MBS and centrifuge at 15,000rpm for 2h at 4°C. Resuspend pellet in MBS, label as "LR," and store at -20°C.
- Collect the stripped membrane pellet from step 5 and resuspend in MBS. Label as "SP" and store at -20°C.
Isolation of PM from Tissue
- Perfuse rat lung as described in Section III,A using steps 1, 2, 3, 8, and 9 only.
- Process rat lung as described in Section III,A except use 3 ml of buffer PM and type "BB" homogenizer vessel for step 2.
- Filter the homogenate through a 53-µm Nytex filter and then through a 30-µM Nytex filter.
- Remove 200µl from the filter solution, label as "homogenate," and store at -20°C. Centrifuge the remaining supernatant at 1000g for 10min at 4°C. Save supernatant on ice. Resuspend the pellet in 3 ml buffer PM and repeat centrifugation. Pool the two supernatants.
- Adjust the volume to 7ml with buffer PM and then add 3ml to make a 30% Percoll solution. Centrifuge at 84,000g for 45min at 4°C (no brakes) in MLS50 rotor. Collect the band around two-thirds to three-fourths from bottom of the tube. Dilute the band material two to three times with PBS and centrifuge at 15,000rpm for 2h at 4°C. Resuspend pellet in 100µl PBS, label as "PM," and store at -20°C.
This protocol is written for T75 flasks, alter proportionately for larger or smaller flasks. All parts of the procedure need to be performed on ice or in a 4°C room.
- Wash cell monolayer three times with cold buffer PM.
- Add 5 ml of cold buffer PM with protease inhibitors (1×). Scrape cells and place in 15-ml centrifuge tube. Centrifuge at 1000g for 5min at 4°C.
- Bring to 1 ml with cold buffer PM with protease inhibitors (1×) (you can use up to six T75 flasks per 1 ml of cold buffer PM with protease inhibitors).
- Place in type "AA" homogenizer vessel. Homogenize with corresponding grinder for 20 strokes at 1800rpm.
- Remove 100 µl, label as "homogenate," and store at -20°C.
- Centrifuge the remaining supernatant at 1000g for 10min at 4°C. Save supernatant on ice. Resuspend the pellet in 1 ml buffer PM and repeat centrifugation. Pool the two supernatants.
- Adjust the volume to 3.5ml with buffer PM and then add 1.5ml to make a 30% Percoll solution. Centrifuge at 84,000g for 45min at 4°C (no brakes) in MLS50 rotor. Collect the band around two-thirds to three-fourths from bottom of the tube. Dilute the band material two to three times with PBS and centrifuge at 15,000rpm for 2h at 4°C. Resuspend pellet in 100µl PBS, label as "PM," and store at -20°C.
Isolation of TRM from Tissue
- Perfuse rat lung as described in Section III,A using steps 1, 2, 3, 8, and 9 only.
- Process rat lung as described in Section III,A except add 1% Triton X-100 to sucrose/HEPES solution in step 2.
- Filter the homogenate through a 53-µM Nytex filter and then through a 30-µM Nytex filter.
- Remove 200µl from the filter solution, label as "homogenate," and store at -20°C. Bring the remaining supernatant to 40% sucrose (w/w)/ 20mM KCl. Layer with discontinuous 35-0% sucrose (w/w)/20mM KCl gradient (3ml each of 35, 30, 25, 20, 15, 10, and 5% sucrose (w/w)/20mM KCl and 20mM KCl as described in Section III,B).
- Centrifuge at 15,000rpm overnight at 4°C in SW28 rotor. Collect the band between 10 and 15% sucrose (w/w) density. Dilute the band material two to three times with PBS and centrifuge at 15,000rpm for 2h at 4°C. Resuspend pellet in PBS, label as "TRM," and store at -20°C.
This protocol is written for T75 flasks; alter proportionately for larger or smaller flasks. All parts of the procedure need to be performed on ice or in a 4°C room.
- Wash cells three times with 1X PBS.
- Scrape cells and place in 15-ml conical tube. Centrifuge at 1000g for 5min at 4°C.
- Resuspend pellet in 1 ml of 1X PBS. Remove 100µl, label this cellular homogenate as "H," and store at -20°C.
- To the remaining H add Triton X-100 to 1% (100 µl of 10% Triton X-100) for a total volume of 1 ml.
- Place in type "AA" homogenizer vessel. Homogenize with corresponding grinder for 20 strokes at 1800rpm.
- Bring to 40% sucrose (w/w)/20mM KCl and layer with discontinuous 35-0% sucrose (w/w)/20mM KCl and centrifuge as described in Section III,B.
- Collect the band between 10 and 15% sucrose (w/w) density. Dilute the band material two to three with PBS and centrifuge at 15,000rpm for 2 h at 4°C. Resuspend pellet in PBS, label as "TRM," and store at -20°C.
- Isolate PM as described in Section III,E.
- Bring pelleted PM to 1 ml with buffer PM.
- Remove 100µl, label as "PM," and store at -20°C.
- To the remaining PM add Triton X-100 to 1% (100µl of 10% Triton X-100) with a total volume of 1 ml. Mix on nutator for 10min at 4°C.
- Bring to 40% sucrose (w/w)/20mM KCl, layer with discontinuous 35-0% sucrose (w/w)/20mM KCl, and centrifuge as described in Section III,B.
- Collect the band between 10 and 15% sucrose (w/w) density. Dilute the band material two to three times with PBS and centrifuge at 15,000rpm for 2h at 4°C. Resuspend pellet in PBS, label as "TRM," and store at -20°C.
- When utilizing cellular membrane subfractions, a rigorous quality control protocol must be applied. It is very important that one first check the purity of the starting material to confirm that the preparation is enriched in markers of the desired membrane material and depleted in markers of other membrane-bound cellular organelles. At the very least, preparations should be checked for markers of the major microdomain components of the plasma membrane, such as caveolae, lipid rafts, adhesion complexes, and the plasma membrane proper, as well as for contaminating intracellular organellar membranes. A good plasma membrane preparation should be enriched 15- to 20-fold in cell surface marker proteins, such as caveolin-1, GPI-AP, integrins, and ion transporters. It should also be depleted by 15- to 20-fold in protein markers expressed in nuclei (ran, transportin), Golgi (p58, β-COP), lysosomes (lamp1), and mitochondria (cytochrome c). Likewise, caveolar preparations must be enriched in caveolar proteins, such as caveolin-1, Gq, and eNOS. LR should be enriched in GPI-AP but lack caveolin-1 expression. In addition, it is also very important to monitor the mass balance of all proteins. For plasma membrane preparations isolated by the silica method, this can be accomplished by examining by Western analysis the proteins left behind in the P-V fraction (containing LR and remaining plasmalemma proper) and comparing them to the protein expression in the P and V fractions to verify that the signal for all three fractions can be accounted for by the fractionation.
- It is very important to maintain the recommended temperatures as presented in these protocols, especially when working with Triton X-100 solubilization. Any increases in temperature may solubilize a greater number of proteins, which may alter the final protein profile.
- One needs to use the correct pH for the solutions, especially those associated with cationic silica particles, as alterations in pH will affect the binding of the silica to the luminal plasma membrane.
- All solutions must be filtered with at least a 0.45-µm filter to ensure that they remain relatively free of contaminants and aggregates.
- Do not mix phosphate solutions with solutions containing colloidal silica, as this will cause the silica to precipitate.
- When perfusing rat organs, be careful to avoid getting air bubbles in the line of the perfusion apparatus, as this will interfere with the vessel lumen-coating procedure and decrease the amount of isolated material.
V. DISCUSSION
The overall goal of these procedures is to excise microdomains specifically from cellular membranes, in this case, from plasma membranes. To isolate any microdomain from a membrane, certain critical criteria are necessary to increase efficiency and purity. First, the microdomain excision must be highly selective. In most cases, however, this cannot be accomplished in one step. One must start with the appropriate highly purified membrane fraction containing the desired microdomain and then process this material to specifically excise and, if necessary, differentially isolate the desired microdomain away from other similarly excised domains.
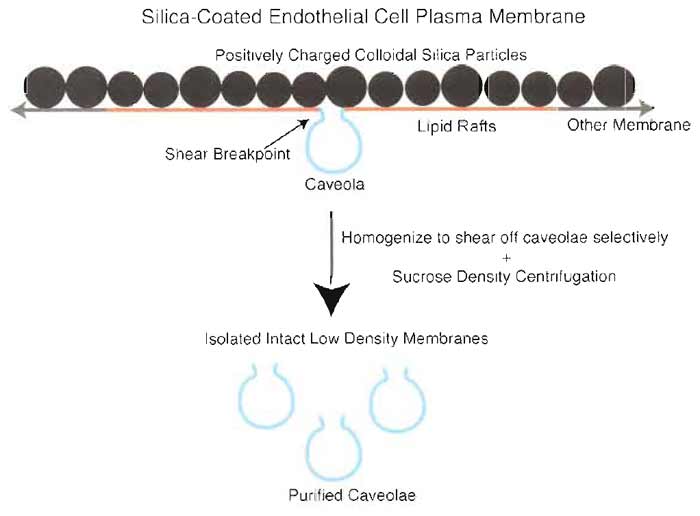 |
| FIGURE 2 Two-step silica coating-based methodology for isolating plasma membrane caveolae. To purify endothelial plasmalemmal caveolae, the luminal endothelial cell membrane is first coated with positively charged colloidal silica particles to create a stable pellicle that specifically marks this membrane and facilitates its purification by density centrifugation of tissue homogenates. These pelleted membranes have many caveolae attached on the side of the membranes opposite to the silica coating that may be stripped from the endothelial membranes by shearing during homogenization at 4°C in the presence of Triton X-100. Further centrifugation through a sucrose density gradient yields a homogeneous population of distinct caveolar vesicles. |
![FIGURE 3 Molecular mapping of luminal plasma membrane subfractions. Proteins (5 µg) from the indicated membrane fractions were subjected to SDS-PAGE followed by electrotransfer to filters for immunoblotting with antibodies to the indicated proteins. H, homogenate; P, silica-coated plasma membrane; PM, percollgradient isolated plasma membranes; V, caveolae [V (in the presence of Triton), V' (in the absence of Triton)], P-V, silica-coated plasma membrane stripped of caveolae [P-V (in the presence of Triton), PV' (in the absence of Triton)]; Tx, Triton-soluble phase; AC, Optiprepisolated, caveolin-enriched membranes; U, material not bound to caveolin antibody-coupled magnetic beads after immunoisolation; B, material bound to caveolin antibody-coupled magnetic beads after immunoisolation.](images/v2_pa_s01_c02_f03.jpg) |
| FIGURE 3 Molecular mapping of luminal plasma membrane subfractions. Proteins (5 µg) from the indicated membrane fractions were subjected to SDS-PAGE followed by electrotransfer to filters for immunoblotting with antibodies to the indicated proteins. H, homogenate; P, silica-coated plasma membrane; PM, percollgradient isolated plasma membranes; V, caveolae [V (in the presence of Triton), V' (in the absence of Triton)], P-V, silica-coated plasma membrane stripped of caveolae [P-V (in the presence of Triton), PV' (in the absence of Triton)]; Tx, Triton-soluble phase; AC, Optiprepisolated, caveolin-enriched membranes; U, material not bound to caveolin antibody-coupled magnetic beads after immunoisolation; B, material bound to caveolin antibody-coupled magnetic beads after immunoisolation. |
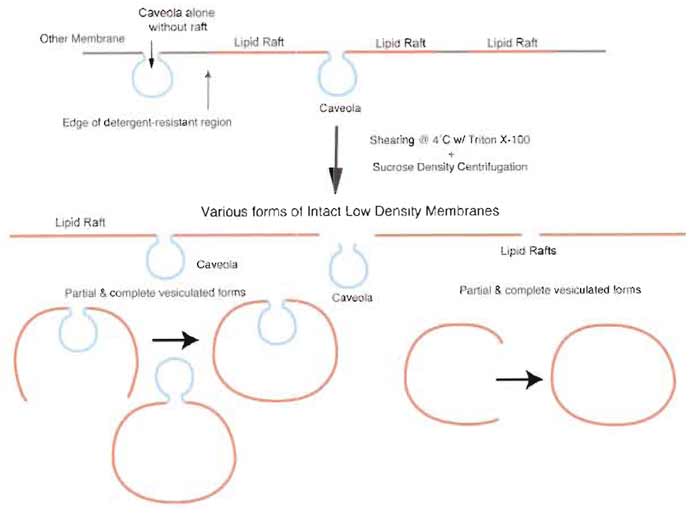 |
| FIGURE 4 Isolation of detergent-resistant membranes without silica-coating procedure yields TRMs containing both caveolae and lipid rafts. Nonsilica-based methodologies that isolate detergent-resistant membranes by sucrose density centrifugation result in a heterogeneous preparation containing a mixture of low-density microdomains as sheets or as partial or fully vesiculated forms. |
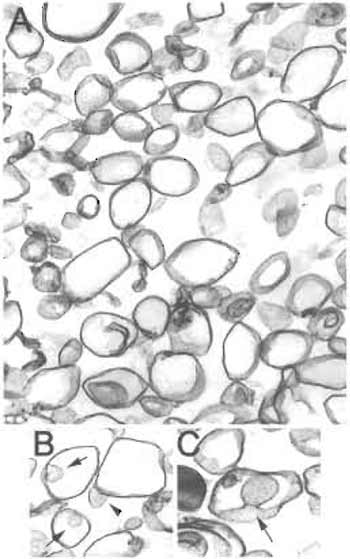 |
| FIGURE 5 Electron microscopy of nonsilica-isolated detergentresistant membranes (TRMs). (A) Membranes isolated without silica consist of larger vesicles (150-700nm diameter) interspersed with smaller caveolar vesicles (<100nm) as well as nonvesiculated linear membrane sheets. (B and C) Caveolae were typically observed attached to a larger vesicle. Arrow: caveola within a larger vesicle. Arrowhead: "inside-out" caveola attached to a larger vesicle. Magnification: A,B = 10,000×; C = 20,000×. |
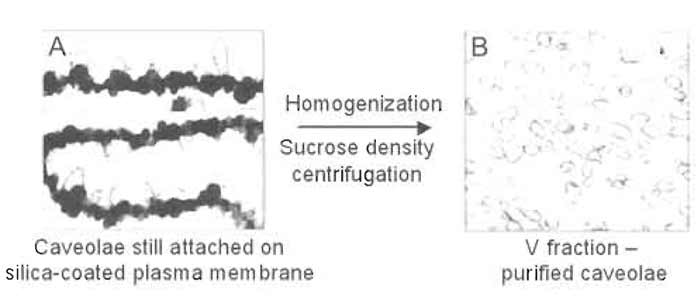 |
| FIGURE 6 Electron micrographs of P (A) and V (B) membrane fractions after isolation. Electron microscopy demonstrates the presence of significant numbers of caveolae on the intracellular side of the plasma membrane not coated with silica (P fraction; A). Mechanical shearing of caveolae from the silicacoated plasma membrane yields a homogeneous population of morphologically distinct caveolae with diameters <90nm (V fraction; B). |
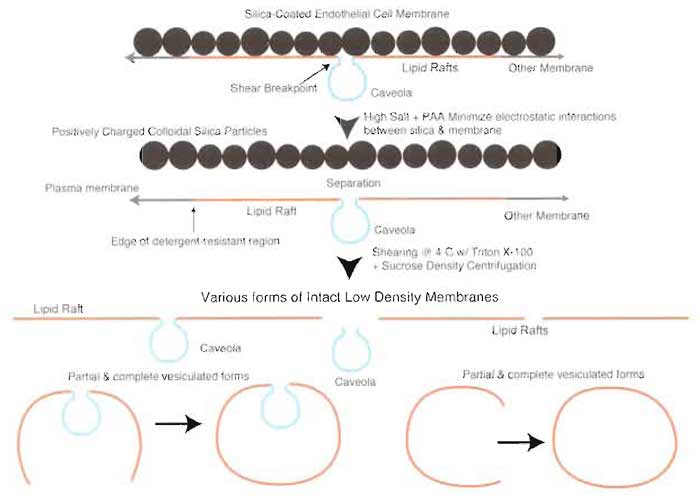 |
| FIGURE 7 Silica coating prevents coisolation of caveolae and lipid rafts. Treatment of silica-coated plasma membranes with high salt prior to separation of caveolae by homogenization results in a heterogeneous mixture of membrane microdomains containing caveolae and lipid rafts alone or attached to each other in partial or fully vesiculated forms. |
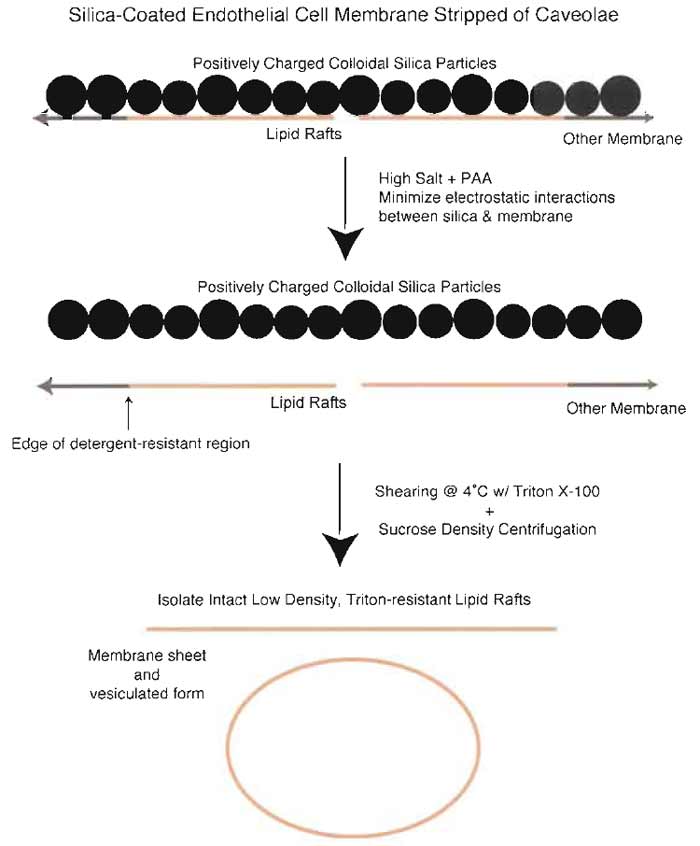 |
| FIGURE 8 Isolation of LR from silica-coated plasma membranes stripped of caveolae. Because the silica coating prevents the release of detergent-resistant, GPI-anchored protein microdomains, it is possible to isolate these domains separately from caveolae. Incubation of silica-coated membranes stripped of caveolae in the presence of high salt and polyacrylic acid followed by homogenization in Triton X-100 and sucrose density centrifugation results in isolation of a membrane fraction containing membrane sheets as well as vesicles >200nm in diameters. These membranes are enriched in GPI-AP, including 5'NT and carbonic anhydrase. |
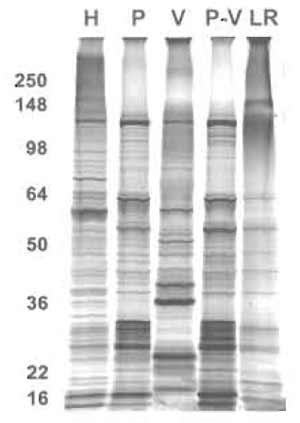 |
| FIGURE 9 One-dimensional protein analysis of various membrane subfraction. Proteins (5µg) from the indicated membrane fractions were subjected to SDS-PAGE followed by silver staining. H, tissue homogenate; P, silica-coated plasma membrane; V, caveolae; P-V, silica-coated plasma membrane stripped of caveolae; LR, lipid rafts. |
A reconstituted cell-free system has also been developed to study further the dynamics and properties of caveolae (Schnitzer et al., 1996). It shows that caveolae are dynamic structures that can be induced to bud from plasma membrane by GTP. Fission of caveolae from the plasmalemma without any physical disruption requires GTP hydrolysis and provides an alternative, and possibly more physiological, source to isolate caveolae. A comparison of GTP-induced budded caveolae with caveolae isolated via the silica-coating technique reveals that both isolates yield similar protein profiles (Schnitzer et al., 1996).
Brown, D. A., and London, E. (2000). Structure and function of sphingolipid- and cholesterol-rich membrane rafts. J. Biol. Chem. 275, 17221-17224.
Brown, D. A., and Rose, J. K. (1992). Sorting of GPI-anchored proteins to glycolipid-enriched membrane subdomains during transport to the apical cell surface. Cell 68, 533-544.
Chang, W. J., Ying, Y. S., Rothberg, K. G., Hooper, N. M., Turner, A. J., Gambliel, H. A., De Gunzburg, J., Mumby, S. M., Gilman, A. G., and Anderson, R. G. (1994). Purification and characterization of smooth muscle cell caveolae. J Cell Biol. 126, 127-138.
Field, K. A., Holowka, D., and Baird, B. (1995). Fc epsilon RImediated recruitment of p53/561yn to detergent-resistant membrane domains accompanies cellular signaling. Proc. Natl. Acad. Sci. USA 92, 9201-9205.
Fra, A. M., Williamson, E., Simons, K., and Parton, R. G. (1995). De novo formation of caveolae in lymphocytes by expression of VIP21-caveolin. Proc. Natl. Acad. Sci. USA 92, 8655-8659.
Fujimoto, T. (1996). GPI-anchored proteins, glycosphingolipids, and sphingomyelin are sequestered to caveolae only after crosslinking. J. Histochem. Cytochem. 44, 929-941.
Gorodinsky, A., and Harris, D. A. (1995). Glycolipid-anchored proteins in neuroblastoma cells form detergent-resistant complexes without caveolin. J. Cell Biol. 129, 619-627.
Liu, J., Oh, P., Horner, T., Rogers, R. A., and Schnitzer, J. (1997). Organized cell surface signal transduction in caveolae distinct from GPI-anchored protein microdomains. J. Biol. Chem. 272, 7211-7222.
Mayor, S., Rothberg, K. G., and Maxfield, E R. (1994). Sequestration of GPI-anchored proteins in caveolae triggered by cross-linking. Science 264, 1948-1951.
McIntosh, D. P., and Schnitzer, J. E. (1999). Caveolae require intact VAMP for targeted transport in vascular endothelium. Am. J. Physiol. 277, H2222-2232.
McIntosh, D. P., Tan, X.-Y., Oh, P., and Schnitzer, J. E. (2002). Targeting endothelium and its dynamic caveolae for tissue-specific transcytosis in vivo: A pathway to overcome cell barriers to drug and gene delivery. Proc. Natl. Acad. Sci. USA 99, 1996-2001.
Monier, S., Parton, R. G., Vogel, E, Behlke, J., Henske, A., and Kurzchalia, T. V. (1995). VIP21-caveolin, a membrane protein constituent of the caveolar coat, oligomerizes in vivo and in vitro. Mol. Biol. Cell 6, 911-927.
Montixi, C., Langlet, C., Bernard, A. M., Thimonier, J., Dubois, C., Wurbel, M. A., Chauvin, J. P., Pierres, M., and He, H. T. (1998). Engagement of T cell receptor triggers its recruitment to lowdensity detergent-insoluble membrane domains. EMBO J. 17, 5334-5348.
Oh, P., McIntosh, D. P., and Schnitzer, J. E. (1998). Dynamin at the neck of caveolae mediates their budding to form transport vesicles by GTP-driven fission from the plasma membrane of endothelium. J. Cell Biol. 141, 101-114.
Oh, P., and Schnitzer, J. E. (1999). Immunoisolation of caveolae with high affinity antibody binding to the oligomeric caveolin cage: Toward understanding the basis of purification [published erratum appears in J. Biol. Chem. 274(41), 29582 (1999)]. J. Biol. Chem. 274, 23144-23154.
Oh, P., and Schnitzer, J. E. (2001). Segregation of heterotrimeric G proteins in cell surface microdomains: Gq binds caveolin to concentrate in caveolae whereas Gi and Gs target lipid rafts by default. Mol. Biol. Cell 12, 685-698.
Razandi, M., Oh, P., Pedram, A., Schnitzer, J., and Levin, E. R. (2002). ERs associate with and regulate the production of caveolin: Implications for signaling and cellular actions. Mol. Endocrinol. 16, 100-115.
Rizzo, V., McIntosh, D. P., Oh, P., and Schnitzer, J. E. (1998). In situ flow activates endothelial nitric oxide synthase in luminal caveolae of endothelium with rapid caveolin dissociation and calmodulin association. J. Biol. Chem. 273, 34724-34729.
Rothberg, K. G., Ying, Y. S., Kolhouse, J. E, Kamen, B. A., and Anderson, R. G. (1990). The glycophospholipid-linked folate receptor internalizes folate without entering the clathrin-coated pit endocytic pathway. J. Cell Biol. 110, 637-649.
Sargiacomo, M., Sudol, M., Tang, Z., and Lisanti, M. P. (1993). Signal transducing molecules and glycosyl-phosphatidylinositol-linked proteins form a caveolin-rich insoluble complex in MDCK cells. J. Cell Biol. 122, 789-807.
Schnitzer, J. E., McIntosh, D. P., Dvorak, A. M., Liu, J., and Oh, P. (1995b). Separation of caveolae from associated microdomains of GPI-anchored proteins. Science 269, 1435-1439.
Schnitzer, J. E., and Oh, P. (1996). Aquaporin-1 in plasma membrane and caveolae provides mercury-sensitive water channels across lung endothelium. Am. J. Physiol. 270, H416-422.
Schnitzer, J. E., Oh, P., Jacobson, B. S., and Dvorak, A. M. (1995c). Caveolae from luminal plasmalemma for rat lung endothelium: Microdomains enriched in caveolin, Ca2+-ATPase and inositol trisphosphate receptor. Proc. Natl. Acad. Sci. USA 92, 1759-1763.
Schnitzer, J. E., Oh, P., and McIntosh, D. P. (1996). Role of GTP hydrolysis in fission of caveolae directly from plasma membrane. Science 274, 239-242.
Shenoy-Scaria, A. M., Kwong, J., Fujita, T., Olszowy, M. W., Shaw, A. S., and Lublin, D. M. (1992). Signal transduction through decay-accelerating factor: Interaction of glycosyl-phosphatidylinositol anchor and protein tyrosine kinases p561ck and p59fyn 1. J. Immunol. 149, 3535-3541.
Smart, E. J., Ying, Y. S., Mineo, C., and Anderson, R. G. (1995). A detergent-free method for purifying caveolae membrane from tissue culture cells. Proc. Natl. Acad. Sci. USA 92, 10104-10108.
Stefanova, I., Horejsi, V., Ansotegui, I. J., Knapp, W., and Stockinger, H. (1991). GPI-anchored cell-surface molecules complexed to protein tyrosine kinases. Science 254, 1016-1019.
Xavier, R., Brennan, T., Li, Q., McCormack, C., and Seed, B. (1998). Membrane compartmentation is required for efficient T cell activation. Immunity 8, 723-732.
Ying, Y. S., Anderson, R. G., and Rothberg, K. G. (1992). Each caveola contains multiple glycosyl-phosphatidylinositol-anchored membrane proteins. Cold Spring Harb. Symp. Quant. Biol. 57, 593-604.
Support our developers




