Isolation of Nucleoli
The human nucleolus is a prominent nuclear substructure assembled around tandemly repeated ribosomal genes (rDNA genes) on chromosomes 13, 14, 15, 21, and 22 and is the site of rDNA transcription and ribosome subunit synthesis (reviewed by Shaw and Jordan, 1995). Visualised under electron microscopy, the nucleolus is separated morphologically into three distinct substructures: fibrillar centres (FC), which are surrounded by dense fibrillar components (DFCs), and granular components (GC) (Shaw and Jordan, 1995). Its high density and structural stability allow effective purification using a straightforward procedure. Following the initial successful attempts to purify nucleoli from human tumour cells and rodent liver cells in the early 1960s (e.g., Muramatsu et al., 1963), numerous studies have reported on the characterization of isolated nucleoli. Nucleoli have been purified from a large variety of mammalian tissues, including liver, brain (Banks and Johnson, 1973), and thyroid (Voets et al., 1979), and from cells of nonmammalian species such as Xenopus (Saiga and Higashinakagawa, 1979) and Tetrahymena (e.g., Matsuura and Higashinakagawa, 1992). The ability to isolate nucleoli in large scale provides an excellent starting material for identifying, purifying, and studying nucleoli and has contributed significantly to the understanding of this nuclear structure.
II. PROCEDURES
Solutions
All solutions are supplemented with Complete protease inhibitor tablet (Roche, Cat. No. 1-873-580) at the final concentration of 1 tablet/50ml).
Phosphate-buffered Saline (PBS)
Buffer A: 10 mM HEPES, pH 7.9, 10 mM KCl, 1.5 mM MgCl2, 0.5 mM dithiothreitol (DTT)
S1 solution: 0.25 M sucrose and 10 mM MgCl2
S2 solution: 0.35 M sucrose, 0.5 mM MgCl2
S3 solution: 0.88 M Sucrose, 0.5 mM MgCl2
See "Notes" on making stock sucrose solution.
- Seed HeLa cells (ATCC number: CCL-2) onto 10 × 14-cm petri dishes and culture at 37°C in 5% CO2 in Dulbecco's modified Eagle medium (DMEM) containing 4mM L-glutamate, 4.5 mg/ml glucose, and 0.11 mg/ml sodium pyruvate (Invitrogen UK, Cat. No: 41966-029), supplemented with 100U/ml penicillin and 100µg/ml streptomycin [1% (v/v) penicillin/ streptomycin solution, Invitrogen UK, Cat. No: 15140- 122] until >90% confluence (approximately 107 cells per dish). This number of HeLa cells consistently provides nucleoli with excellent yield and purity. It is possible to scale down the preparation, although the purity of isolated nucleoli may suffer. Make sure you monitor every step using a phase-contrast microscope (see later). One hour before nucleolar isolation, replace with fresh, prewarmed medium.
- Harvest cells by trpysinization. Rinse each dish three times with prewarmed PBS and, on removal of the last rinse, add 2ml of trypsin-EDTA solution (Invitrogen UK, Cat. No: 25300-054) per dish. Swirl the dishes to make sure the trypsin-EDTA is distributed evenly and return the dishes to the incubator for about 5min. Check under a phase-contrast microscope that all the cells are detached. Prolong incubation if needed. Into each dish add 8 ml of prewarmed medium and pipette up and down so that all the cells are collected as a single-cell suspension. Pool all the harvested cells into 2 × 50-ml Falcon tubes. For some strains of HeLa cells, it is also possible to harvest the cells by scraping them in 5 ml ice-cooled PBS per dish. Because scraping may lead to an impure nucleolar preparation in some HeLa strains, it is not recommended as the method of first choice.
- Wash three times with ice-cold PBS at 218g (1000rpm, Beckman GS-6 centrifuge, GH-3.8 rotor) at 4°C.
- After the final PBS wash, resuspend the cells in 5 ml of buffer A and incubate the cells on ice for 5 min. Put a small drop of the cell suspension on a glass slide and check under a phase-contrast microscope, such as a Zeiss Axiovert 25, using a 20× objective. The cells should be swollen, but not burst (Fig. 1). Nucleoli of cultured mammalian cells disassemble at 37°C in hypotonic conditions (Zatsepina et al., 1997). It is therefore imperative to keep the cell suspension on ice during this step.
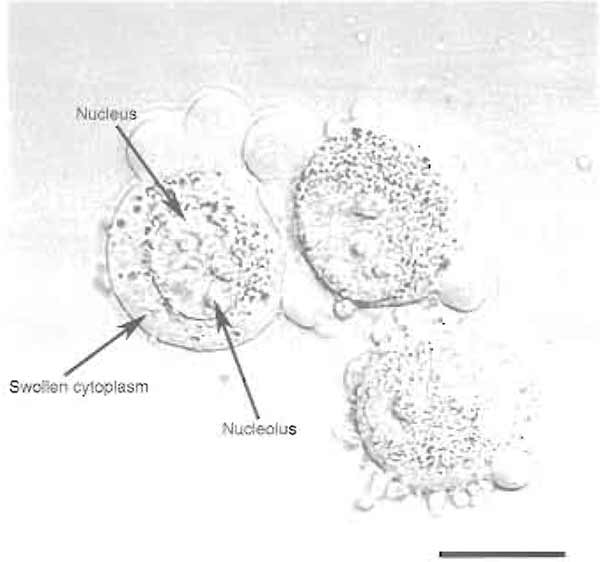
FIGURE 1 HeLa cells after step 4. Note the swollen cytoplasm and prominent nucleoli. Bar: 10 µm.
- Transfer the cell suspension to a precooled 7-ml Dounce tissue homogenizer (Wheaton Scientific Product Cat. No: 357542). Homogenize 10 times using a tight pestle ("A" specification: 0.0010-0.0030-in. clearance) while keeping the homogenizer on ice. The number of strokes needed depends on the cell type used (see Section III). It is therefore necessary to check the homogenized cells under a phase-contrast microscope after every 10 strokes. Stop when >90% of the cells are burst, leaving intact nuclei, with various amounts of cytoplasmic material attached. In most cases, the presence of this cytoplasmic contamination does not affect the final purity of the isolated nucleoli (Fig. 2). Centrifuge the homogenized cells at 218g (1000rpm, Beckman GS-6 centrifuge, GH-3.8 rotor) for 5min at 4°C. The pellet contains enriched, but not highly pure, nuclei.
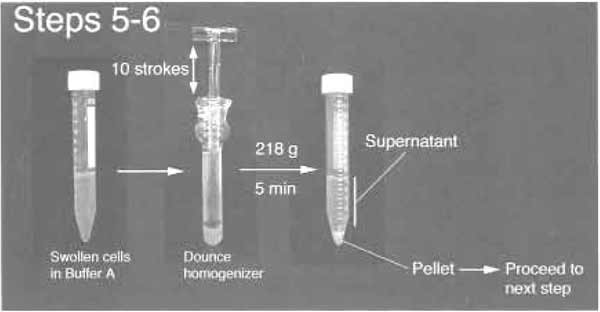
FIGURE 2 Step 5 and 6 of the procedure.
- Resuspend the pellet with 3 ml S1 solution (Fig. 3). The pellet should be resuspended readily by pipetting up and down. A pellet that cannot be resuspended contains lysed nuclei and should be discarded. Layer the resuspended pellet over 3ml of the S2 solution. Take care to keep the two layers cleanly separated. Centrifuge at 1430g (2500rpm, Beckman GS-6 centrifuge, GH-3.8 rotor) for 5 min at 4°C. This step results in a cleaner nuclear pellet (Fig. 3). Resuspend the pellet with 3 ml of the S2 solution by pipetting up and down.
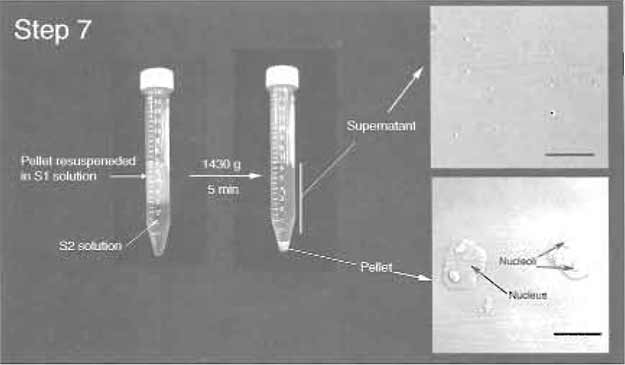
FIGURE 3 Step 7 of the procedure. Note the clear boundary between S1 and S2 layers before centrifugation. (Insets) DIC images of the supernatant and pellet. Note prominent nucleoli inside nuclei in the pellet. Bars: 10 µm.
- Sonicate the nuclear suspension with six 10-s bursts (with 10-s intervals between each burst) using a Misonix XL 2020 sonicator fitted with a microtip probe and set at power setting 5 (Fig. 4A). Check the sonicated nuclei under a phase-contrast microscope. There should be virtually no intact cells and nucleoli should be readily observed as dense, refractile bodies (Fig. 4B). The optimal sonication time depends on the cell type used. If you attempt to isolate nucleoli from a cell type from the first time, it is necessary to check the sonicated material under a microscope after every 10sec of sonication. Oversonication leads to destruction of nucleoli.
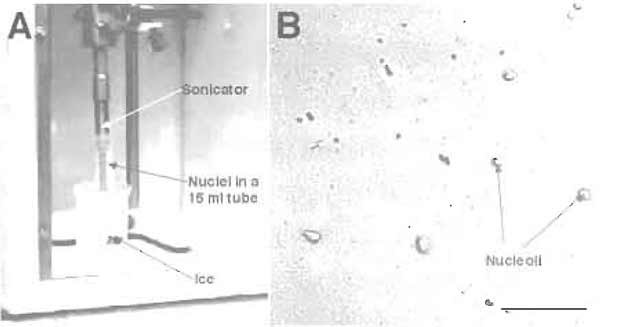
FIGURE 4 (A) Setup for sonication. (B) DIC image of sonicated nuclei. Note the presence of prominent nucleoli. Bar: 10 µm.
- Layer the sonicated sample over 3 ml of the S3 solution and centrifuge at 3000g (3500rpm, Beckman GS-6 centrifuge, GH-3.8 rotor) for 10min at 4°C (Fig. 5). The pellet contains nucleoli, whereas the supernatant can be retained as the "nucleoplasmic fraction" (Fig. 5).
- Resuspend the nucleoli with 0.5 ml of the S2 solution, followed by centrifugation at 1430g (2500rpm, Beckman GS-6 centrifuge, GH-3.8 rotor) for 5min at 4°C. The pellet contains highly purified nucleoli. Check under a phase-contrast microscope to ensure that this preparation contains only highly purified nucleoli without any other material (Fig. 5). Nucleoli can be resuspended in 0.5ml of the S2 solution and stored at -80°C.
 |
| FIGURE 5 Step 9 of the procedure. Note the clear jboundary between S2 and S3 layers before and after centrifugation. The pellet should be small but visible. (Insets) DIC images of the supernatant and pellet. The pellet should contain purified nucleoli. Bars: 10µm (left inset) and 20µm (right inset). |
1. Making 2.55 M Sucrose Stock
Here is a protocol for preparing a sucrose stock solution (Cline and Ryel, 1971) suitable for the nucleolar isolation protocol. The resulting solution is 2.55M, or 66% by weight. Its density is 1.3224g/cm3 at 20°C, and its refractive index is 1.4558. The stock solution is stable indefinitely at 4°C. This procedure can be carried out at room temperature. There is no need to heat up the solution to help dissolve the sucrose. Heating up an incompletely dissolved sucrose solution can lead to charring of sucrose and affect the quality of the sucrose solution.
- Weigh out 1710g sucrose (BDH). Keep it aside in a clean container.
- Put exactly 900ml water and a magnetic bar in a 5 litre beaker. Put the beaker on a stirrer and start stirring.
- Add one-third of the sucrose into the beaker. Make sure the magnetic bar is rotating freely. Stir for 1 h.
- Add another one-third of the sucrose into the solution. Again make sure the rotation of the stir bar is not impaired. Stir for another 1 h.
- Add the remaining sucrose. Stir for another 1h or until all the sucrose has gone into solution. The final volume should be exactly 2 litres.
We use a Misonix 2020 sonicator fitted with a microtip at power setting 5. To ensure reproducible soncation, the following points should be followed.
- It is necessary to tune the sonicator every time after you change the probe. Follow the manufacturer's manual for the tuning procedure.
- Sonication produces intense and localized heat in your solution. If you are concerned about the heating, the correct way to reduce heating is to shorten the sonication time and to increase the intermission between bursts. Keeping the tube on ice or performing the sonication in the cold room is helpful, but is not the most effective way of heat control.
- If the probe is too close to the liquid surface, it produces a foam and reduces the efficiency of sonication. Make sure the probe is well submerged in the solution, about 5 mm above the bottom of the tube. Do not, however, touch the bottom or the wall of the tube with the probe.
- A sonicator probe that has been used repeatedly develops pits on its end. The sonication efficiency gradually decreases as time goes on. Therefore, the sonication time recommended here can only be used as a guideline. Always monitor the outcome of sonication using a phase-contrast microscope. You may need to adjust the sonication time to maintain the efficiency, especially if the probe is getting old. Change the probe when the efficiency is noticeably down.
- To immunolabel purified nucleoli, spot about 5 µl of the nucleolar suspension onto a polylysine-coated slide (BDH Cat. No: 406/0178/00) and air dry the spot. Rehydrate the slide in PBS for 5min before carrying out a standard immunostaining procedure.
- To separate nucleolar proteins on a gel, resuspend directly either in Laemmli SDS sample buffer or in your preferred buffer. The high concentration of nucleic acid in isolated nucleoli makes the lysed sample very viscous. The sample can be clarified by passing through a QIAshredder spin column (Qiagen Cat. No: 79654). Nucleoli can also be extracted with RIPA buffer (150 mM NaCl, 1% NP40, 0.5% deoxycholate, 0.1% SDS, 50mM Tris, pH 8.0, Complete protease inhibitor cocktail). Immunoprecipitations can be performed from nucleolar lysates prepared in RIPA buffer.
4. Adapting Nucleolar Isolation Protocol to Use with Other Cell Types
The aforementioned protocol can be adapted readily to other cell types. Apart from HeLa cells, we have used this protocol, with minor modifications, to isolated nucleoli from MCF-7 (human breast epithelium), WI-38 (human fibroblast), IMR-32 (human neuroblastoma), HL60 (human promyelocytic leukemia), and plant Arabidopsis thalina cells. When adapting the protocol to a different cell type, make sure that you control each step by carefully checking the products after each step under a phase-contrast microscope. For example, different cell types may require a different homogenization (step 4) and/or sonication strength (step 7). The concentration of MgCl2 also appears crucial to the purity of the isolated nucleoli. If the isolated nucleoli are not pure enough, try lowering the concentration of MgCl2 in the S2 and S3 solutions. If the yield is poor or if nucleoli look fragmented, use more MgCl2.
Andersen, J. S., Lyon, C. E., Fox, A. H., et al. (2002). Directed proteomic analysis of the human nucleolus. Curt. Biol. 12, 1-11.
Banks, S. P., and Johnson, T. C. (1973). Developmental alterations in RNA synthesis in isolated mouse brain nucleoli. Biochim. Biophys. Acta 294, 450-460.
Cline, G. B., and Ryel, R. B. (1971). Methods Enzymol. 22, 168-204.
Matsuura, T., and Higashinakagawa, T. (1992). In vitro transcription in isolated nucleoli of Tetrahymena pyriformis. Dev. Genet. 13, 143-150.
Muramatsu, M., Smetana, K., and Busch, H. (1963). Quantitative aspects of isolation of nucleoli of the Walker carcinosarcoma and liver of the rat. Cancer Res. 25, 693-697.
Shaw, P. J., and Jordan, E. G. (1995). The nucleolus. Annu. Rev. Cell Dev. Biol. 11, 93-121.
Voets, R., Lagrou, A., Hilderson, H., et al. (1979). RNA synthesis in isolated bovine thyroid nuclei and nucleoli: alpha-amanitin effect, a hint to the existence of a specific regulatory system. Hoppe Seylers Z Physiol. Chem. 360, 1271-1283.
Zatsepina, O. V., Dudnic, O. A., Chentsov, Y. S., Thiry, M., Spring, H., and Trendelenburg, M. E (1997). Reassembly of functional nucleoli following in situ unraveling by low-ionic-strength treatment of cultured mammalian cells. Exp. Cell. Res. 233(1), 155-168.
Support our developers




