Phenylethylamines and Simple Tetrahydroisoquinoline Alkaloids
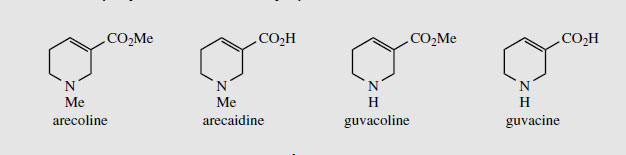
Areca Areca nuts (betel nuts) are the seeds of Areca catechu (Palmae/Arecaceae), a tall palm cultivated in the Indian and Asian continents. These nuts are mixed with lime, wrapped in leaves of the betel pepper (Piper betle) and then chewed for their stimulant effect, and subsequent feeling of well-being and mild intoxication. The teeth and saliva of chewers stain bright red. The major stimulant alkaloid is arecoline (up to 0.2%) (Figure 36), the remainder of the alkaloid content (total about 0.45%) being composed of related reduced pyridine structures, e.g. arecaidine, guvacine (tetrahydronicotinic acid), and guvacoline (Figure 36). Arecoline is an agonist for muscarinic acetylcholine receptors (Figure 34), although it possesses a reversed ester profile compared with acetylcholine. Arecolinehas been employed in veterinary practice as a vermicide to eradicate worms. |
 |
| Figure 36 |
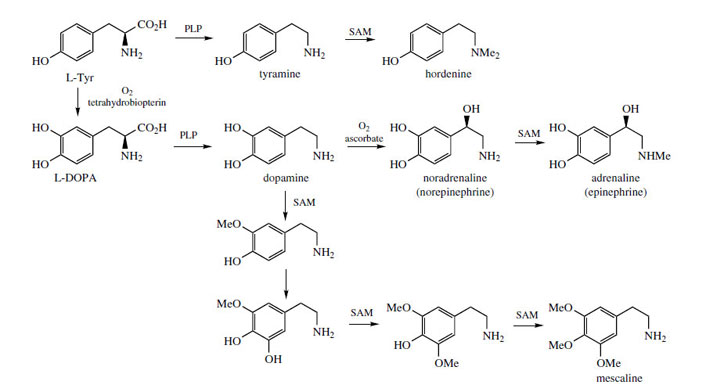
PLP-dependent decarboxylation of L-tyrosine gives the simple phenylethylamine derivative tyramine, which on di-N-methylation yields hordenine, a germination inhibitory alkaloid from barley (Hordeum vulgare; Graminae/ Poaceae) (Figure 37). More commonly, phenylethylamine derivatives possess 3,4-di- or 3,4,5-tri-hydroxylation patterns, and are derived via dopamine (Figure 37), the decarboxylation product from L-DOPA (L-dihydroxyphenylalanine). Pre-eminent amongst these are the catecholamines* noradrenaline (norepinephrine), a mammalian neurotransmitter, and adrenaline (epinephrine), the ‘fight or flight’ hormone released in animals from the adrenal gland as a result of stress. These compounds are synthesized by successive β-hydroxylation and N-methylation reactions on dopamine (Figure 37). Aromatic hydroxylation and O-methylation reactions in the cactus Lophophora williamsii* (Cactaceae) convert dopamine into mescaline (Figure 37), an alkaloid with pyschoactive and hallucinogenic properties. Note that the sequence of hydroxylations and methylations exactly parallel those described for the cinnamic acids.
Catecholamines
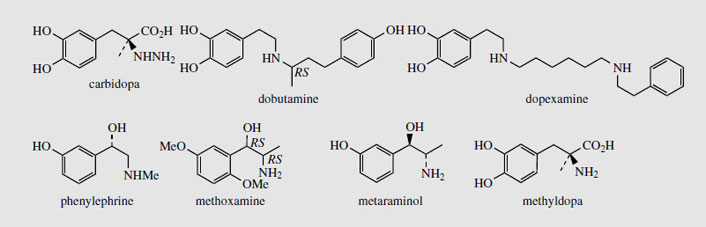
The catecholamines dopamine, noradrenaline (norepinephrine), and adrenaline (epinephrine) are produced in the adrenal glands and nervous tissue and act as neurotransmitters in mammals. Several adrenergic receptors have been identified. α-Receptors are usually excitatory and produce a constricting effect on vascular, uterine, and intestinal muscles. β-Receptors are usually inhibitory on smooth muscle, but stimulatory on heart muscles. Dopamine (Figure 37) can act on both vascular α1 and cardiac β1 receptors, but also has its own receptors in several other structures. In Parkinson's disease, there is a deficiency of dopamine due to neural degeneration, affecting the balance between excitatory and inhibitory transmitters. Treatment with L-DOPA (levodopa) (Figure 37) helps to increase the dopamine levels in the brain. Unlike dopamine, DOPA can cross the blood-brain barrier, but needs to be administered with a DOPA-decarboxylase inhibitor, e.g. carbidopa (Figure 38), to prevent rapid decarboxylation in the bloodstream. Injections of dopamine or dobutamine (Figure 38) are valuable as cardiac stimulants in cases of cardiogenic shock. These agents act on β1 receptors; dopexamine(Figure 38) is also used for chronic heart failure but acts on β2 receptors in cardiac muscle.
Noradrenaline (norepinephrine) (Figure 37) is a powerful peripheral vasoconstrictor predominantly acting on α-adrenergic receptors, and is useful in restoring blood pressure in cases of acute hypotension. The structurally related alkaloid ephedrine may be used in the same way, and synthetic analogues of noradrenaline, e.g. phenylephrine, methoxamine, and metaraminol (Figure 38), have also been developed. Methyldopais used to treat hypertension; it is a centrally acting agent that becomes decarboxylated and hydroxylated to form the false transmitter α-methylnoradrenaline, which competes with noradrenaline.
 |
| Figure 37 |
 |
| Figure 38 |
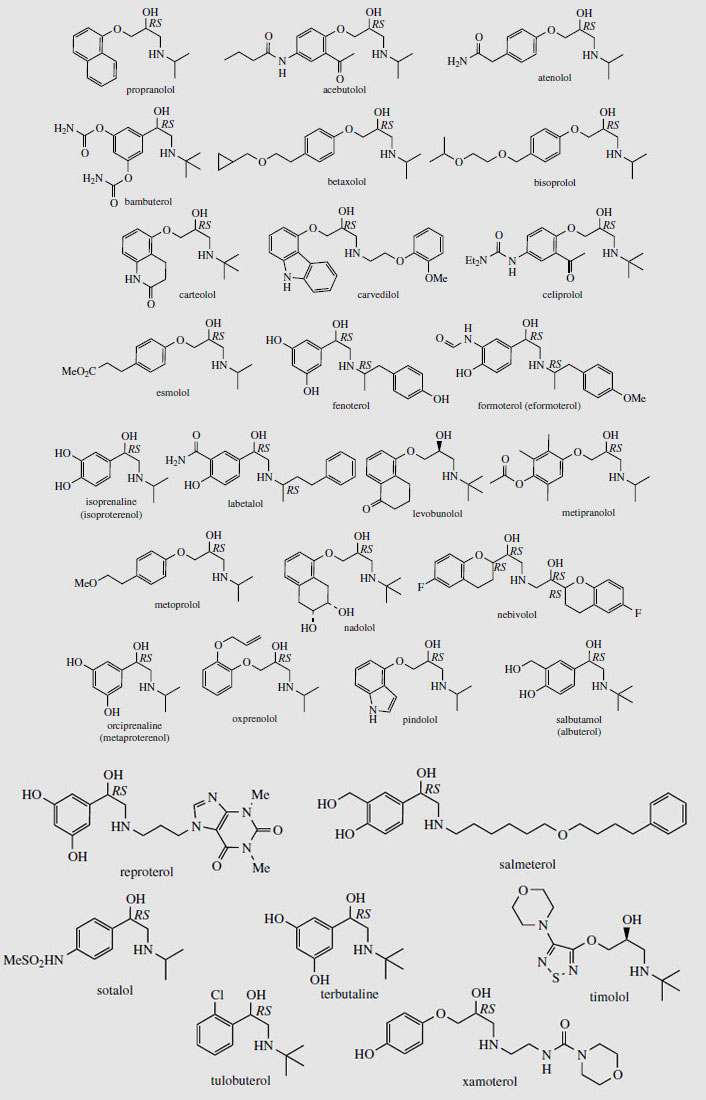
Adrenaline (epinephrine) (Figure 37) is released from the adrenal glands when an animal is confronted with an emergency situation, markedly stimulating glycogen breakdown in muscle, increasing respiration, and triggering catabolic processes that result in energy release. Adrenaline interacts with both α- and β- receptors, an a-response being vasoconstriction of smooth muscle in the skin. β-Responses include mediation of cardiac muscle contractions and the relaxation of smooth muscle in the bronchioles of the lung. Injection of adrenaline is thus of value in cases of cardiac arrest, or in allergic emergencies such as bronchospasm or severe allergy (anaphylactic shock). It is not effective orally. A wide range of cardioactive β-adrenoceptor blocking agents (beta-blockers) has been developed to selectively bind to β-receptors to control the rate and force of cardiac contractions in the management of hypertension and other heart conditions. The prototype of the beta-blocker drugs is propranolol (Figure 39), in which the catechol ring system has been modified to a naphthalene ether, and a bulky N-alkyl substituent has been incorporated. Many structural variants have been produced and there is now a huge, perhaps bewildering, variety of beta-blockers in regular use, with subtle differences in properties and action affecting the choice of drug for a particular condition or individual patient. These are shown in Figure 39. Atenolol, betaxolol, bisoprolol, metoprolol, nebivolol, and to a lesser extent acebutolol, have less effect on the β2 bronchial receptors and are thus relatively cardioselective. Most other agents are non-cardioselective, and could also provoke breathing difficulties. Esmolol and sotalol are used only in the management of arrhythmias.
 |
| Figure 39 |
Other β-agonists are valuable as antiasthmatic drugs. Important examples include salbutamol (albuterol) and terbutaline, which are very widely prescribed, principally for administration by inhalation at the onset of an asthma attack, but, as with cardioactive beta-blockers, a wide range of agents is in current use (Figure 39). These agents are mainly selective towards the β2-receptors, and supersede the earlier less selective bronchodilator drugs such as isoprenaline (isoproterenol) and orciprenaline(metaproterenol) (Figure 39). Topical application of a beta-blocker to the eye reduces intraocular pressure by reducing the rate of production of aqueous humour. Some drugs in this class, namely betaxolol, carteolol, levobunolol, metipranolol, and timolol, are thus useful in treating glaucoma. Propranolol, metoprolol, nadolol, and timolol also have additional application in the prophylaxis of migraine.
Catecholamine neurotransmitters are subsequently inactivated by enzymic methylation of the 3-hydroxyl (via catechol-O-methyltransferase) or by oxidative removal of the amine group via monoamine oxidase. Monoamine oxidase inhibitors are sometimes used to treat depression, and these drugs cause an accumulation of amine neurotransmitters. Under such drug treatment, simple amines such as tyramine in cheese, beans, fish, and yeast extracts are also not metabolized and can cause dangerous potentiation of neurotransmitter activity.
Lophophora
Lophophora or peyote consists of the dried sliced tops of Lophophora williamsii (Cactaceae), a small cactus from Mexico and the SW United States. The plant has been used by the Aztecs and since by the Mexican Indians for many years, especially in religious ceremonies to produce hallucinations and establish contact with the gods. The so-called mescal buttons were ingested and this caused unusual and bizarre coloured images. The plant is still used by people seeking drug-induced experiences. The most active of the range of alkaloids found in lophophora (total 8-9% alkaloids in the dried mescal buttons) is mescaline (Figure 37), a simple phenylethylamine derivative. Other constituents include anhalamine, anhalonidine, and anhalonine (Figure 40). Mescaline has been used as a hallucinogen in experimental psychiatry. The dosage required is quite large (300-500 mg), but the alkaloid can readily be obtained by total synthesis, which is relatively uncomplicated. Mescaline is also found in other species of cactus, e.g. Trichocereus pachanoi, a substantially larger columnar plant that can grow up to 20 feet tall, and found mainly in the Andes.
Closely-related alkaloids cooccurring with mescaline are anhalamine, anhalonine, and anhalonidine (Figure 40), which are representatives of simple tetrahydroisoquinoline derivatives. The additional carbon atoms, two in the case of anhalonidine and anhalonine, and one for anhalamine, are supplied by pyruvate and glyoxylate respectively. In each case, a carboxyl group is lost from this additional precursor. The keto acid pyruvate reacts with a suitable phenylethylamine, in this case the dimethoxy-hydroxy derivative, giving a Schiff base (Figure 40). In a Mannichlike mechanism, cyclization occurs to generate the isoquinoline system, the mesomeric effect of an oxygen substituent providing the nucleophilic site on the aromatic ring. Restoration of aromaticity via proton loss gives the tetrahydroisoquinoline, overall a biosynthetic equivalent of the Pictet–Spengler synthesis. The carboxyl group is then removed, not by a simple decarboxylation, but via an unusual oxidative decarboxylation first generating the intermediate imine, reduction finally leading to anhalonidine with further methylation giving anhalonine. Anhalamine is derived from the same phenylethylamine precursor utilizing glyoxylic acid (Figure 40).
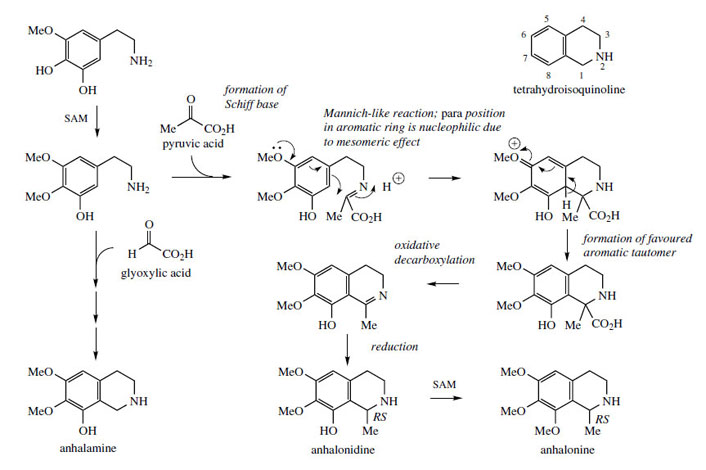 |
| Figure 40 |
The chemical synthesis of tetrahydroisoquinolines by the Pictet–Spengler reaction does not usually employ keto acids like pyruvate or aldehyde acids like glyoxylate. Instead, simple aldehydes, e.g. acetaldehyde or formaldehyde, could be used (Figure 41, a), giving the same product directly without the need for a decarboxylation step to convert the intermediate tetrahydroisoquinolinecarboxylic acid (Figure 41, b). In nature, both routes are in fact found to operate, depending on the complexity of the R group. Thus, the keto acid (route b) is used for relatively simple substrates (R = H, Me) whilst more complex precursors (R = ArCH2, ArCH2 CH2, etc) are incorporated via the corresponding aldehydes (route a). The stereochemistry in the product is thus controlled by the condensation/Mannich reactions (route a), or by the final reduction reaction (route b). Occasionally, both types of transformation have been demonstrated in the production of a single compound, an example being the Lophophora schotti alkaloid lophocerine (Figure 42).
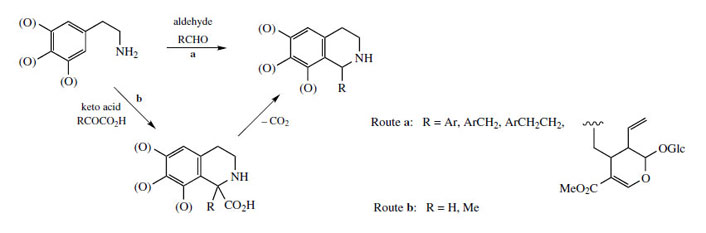 |
| Figure 41 |
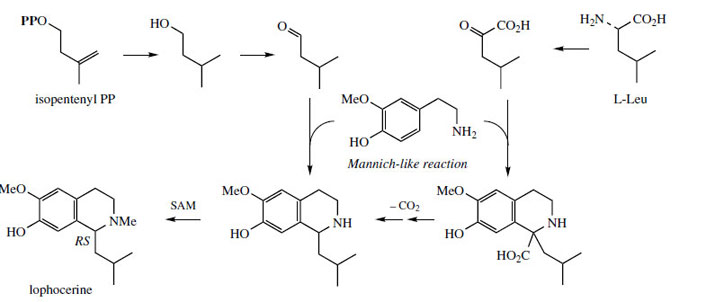 |
| Figure 42 |
This requires utilization of a C5 isoprene unit, incorporated via an aldehyde. However, a second route using the keto acid derived from the amino acid L-leucine by transamination has also been demonstrated. The alkaloid salsolinol (Figure 43) is found in plants, e.g. Corydalis spp. (Papaveraceae), but can also be detected in the urine of humans as a product from dopamine and acetaldehyde combining via a Pictet–Spengler reaction. Acetaldehyde is typically formed after ingestion of ethanol.
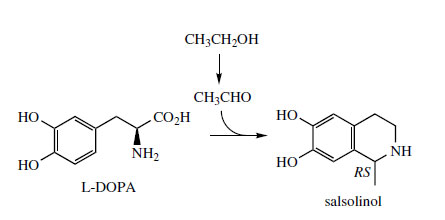 |
| Figure 43 |
 |
| Figure 44 |
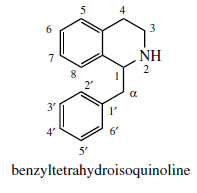
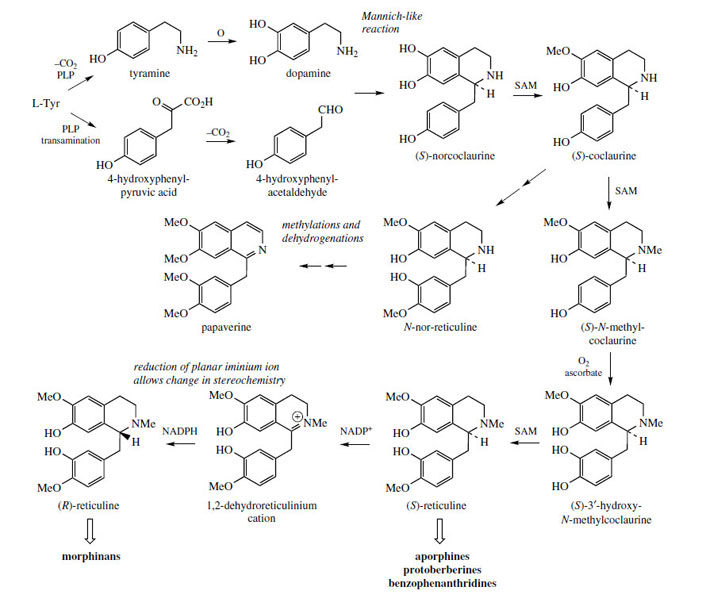
Incorporation of a phenylethyl unit into the phenylethylamine gives rise to a benzyltetrahydroisoquinoline skeleton (Figure 44), which can undergo further modifications to produce a wide range of plant alkaloids, many of which feature as important drug materials. Fundamental changes to the basic skeleton increase the diversity of structural types as described under ‘modified benzyltetrahydroisoqinolines’. Most examples of benzyltetrahydroisoquinoline alkaloids and modified structures contain ortho di-oxygenation in each aromatic ring, which pattern is potentially derivable from the utilization of two DOPA molecules. Although two tyrosine molecules are used in the biosynthetic pathway, only the phenylethylamine fragment of the tetrahydroisoquinoline ring system is formed via DOPA, the remaining carbons coming from tyrosine via 4-hydroxyphenylpyruvic acid and 4-hydroxyphenylacetaldehyde (Figure 45). The product from the Mannich-like reaction is thus the trihydroxy alkaloid norcoclaurine, formed stereospecifically as the (S )-enantiomer. The tetrahydroxy substitution pattern is built up by further hydroxylation in the benzyl ring, though O-methylation [giving (S )-coclaurine] and N-methylation steps precede this. Eventually, (S )-reticuline, a pivotal intermediate to other alkaloids, is attained by N-methylation. Surprisingly, some alkaloids, such as the opium alkaloids morphine, codeine, and thebaine are elaborated from (R)-reticuline rather than the first-formed (S )-isomer. The change in configuration is known to be achieved by an oxidation–reduction process and the intermediate1,2-dehydroreticulinium ion, as shown in Figure 45. Papaverine, a benzylisoquinoline alkaloid found in opium, is formed from N-nor-reticuline by successive Omethylations and oxidation in the heterocyclic ring (Figure 45).
 |
| Figure 45 |
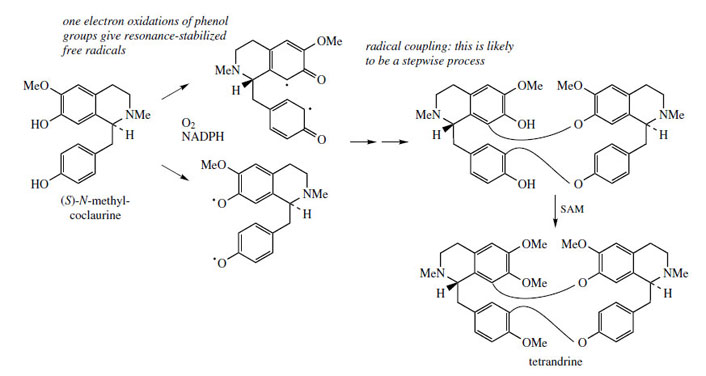
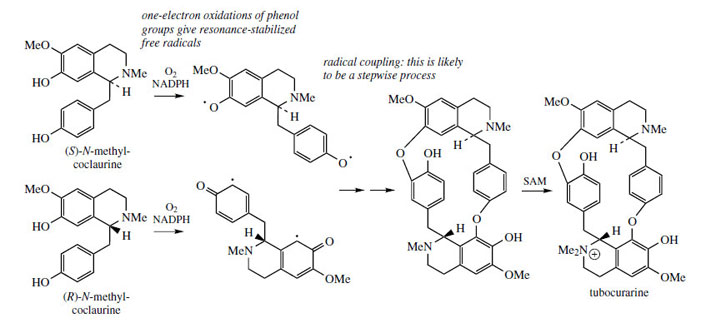
Structures in which two (or more) benzyltetrahydroisoquinoline units are linked together are readily explained by a phenolic oxidative coupling mechanism. Thus, tetrandrine (Figure 46), a bis-benzyltetrahydroisoquinoline alkaloid isolated from Stephania tetrandra (Menispermaceae) is easily recognized as a coupling product from two molecules of (S )-N-methylcoclaurine (Figure 46). The two diradicals, formed by oneelectron oxidations of a free phenol group in each ring, couple to give ether bridges, and the product is then methylated to tetrandrine. The pathway is much more likely to follow a stepwise coupling process requiring two oxidative enzymes rather than the combined one suggested in Figure 46. Tetrandrine is currently of interest for its ability to block calcium channels, and may have applications in the treatment of cardiovascular disorders. By a similar mechanism, tubocurarine (Figure 47), the principal active component in the arrow poison curare* from Chondrodendron tomentosum (Menispermaceae), can be elaborated by a different coupling of one molecule each of (S )- and (R)-N-methylcoclaurine (Figure 47).
 |
| Figure 46 |
 |
| Figure 47 |
Curare
Curare is the arrow poison of the South American Indians, and it may contain as many as 30 different plant ingredients, which may vary widely from tribe to tribe according to local custom. Curare is prepared in the rain forests of the Amazon and Orinoco, and represents the crude dried extract from the bark and stems of various plants. The young bark is scraped off, pounded, and the fibrous mass percolated with water in a leaf funnel. The liquor so obtained is then concentrated by evaporation over a fire. Further vegetable material may be added to make the preparation more glutinous so that it will stick to the arrows or darts. The product is dark brown or black, and tarlike.
In the 1880s, it was found that the traditional container used for curare was fairly indicative of the main ingredients that had gone into its preparation. Three main types were distinguished. Tube curare was packed in hollow bamboo canes, and its principal ingredient was the climbing plant Chondrodendron tomentosum (Menispermaceae). Calabash curare was packed in gourds, and was derived from Strychnos toxifera (Loganiaceae). Pot curare was almost always derived from a mixture of loganiaceous and menispermaceous plants, and was packed in small earthenware pots. Current supplies of curare are mainly of the menispermaceous type, i.e. derived from Chondrodendron.
The potency of curare as an arrow poison is variable and consequently needs testing. A frequently quoted description of this testing is as follows: 'If a monkey hit by a dart is only able to get from one tree to the next before it falls dead, this is ''one-tree curare'', the superior grade. ''Two-tree curare'' is less satisfactory, and ''three-tree curare'' is so weak that it can be used to bring down live animals that the Indians wish to keep in captivity.' Thus, the poison does not necessarily cause death; it depends on the potency. Curare is only effective if it enters the bloodstream, and small amounts taken orally give no ill effects provided there are no open sores in the mouth or throat.
Curare kills by producing paralysis, a limp relaxation of voluntary muscles. It achieves this by competing with acetylcholine at nicotinic receptor sites, thus blocking nerve impulses at the neuromuscular junction. Death occurs because the muscles of respiration cease to operate, and artificial respiration is an effective treatment prior to the effects gradually wearing off through normal metabolism of the drug. Anticholinesterase drugs such as physostigmine and neostigmine are specific antidotes for moderate curare poisoning. Curare thus found medicinal use as a muscle relaxant, especially in surgical operations such as abdominal surgery, tonsillectomy, etc, where tense muscles needed to be relaxed. Curare was also found to be of value in certain neurological conditions, e.g. multiple sclerosis, tetanus, and Parkinson's disease, to temporarily relax rigid muscles and control convulsions, but was not a curative. However, the potency of curare varied markedly, and supplies were sometimes limited.
The alkaloid content of curare is from 4% to 7%. The most important constituent in menispermaceous curare is the bis-benzyltetrahydroisoquinoline alkaloid (+)-tubocurarine (Figure 48). This is a monoquaternary ammonium salt, and is water soluble. Other main alkaloids include non-quaternary dimeric structures, e.g. isochondrodendrine and curine (bebeerine) (Figure 48), which appear to be derived from two molecules of (R)-Nmethylcoclaurine, with the former also displaying a different coupling mode. The constituents in loganiaceous curare (from calabash curare, i.e. Strychnos toxifera) are even more complex, and a series of 12 quaternary dimeric strychnine-like alkaloids has been identified, e.g. C-toxiferine (toxiferine-1).
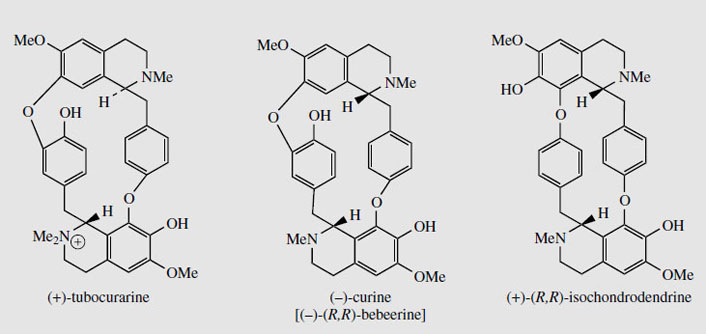 |
| Figure 48 |
Tubocurarine (Figure 48) is still extracted from menispermaceous curare and injected as a muscle relaxant in surgical operations, reducing the need for deep anaesthesia. Artificial respiration is required until the drug has been inactivated (about 30 minutes) or antagonized (e.g. with neostigmine). The limited availability of tubocurarine has led to the development of a series of synthetic analogues, some of which have improved characteristics and are now preferred over the natural product. Interestingly, the structure of tubocurarine was originally formulated incorrectly as a diquaternary salt, rather than the monoquaternary salt, and analogues were based on the pretext that curare-like effects might be obtained from compounds containing two quaternary nitrogens separated by a polymethylene chain. This was borne out in practice, and the separation was found to be optimal at about ten carbons.
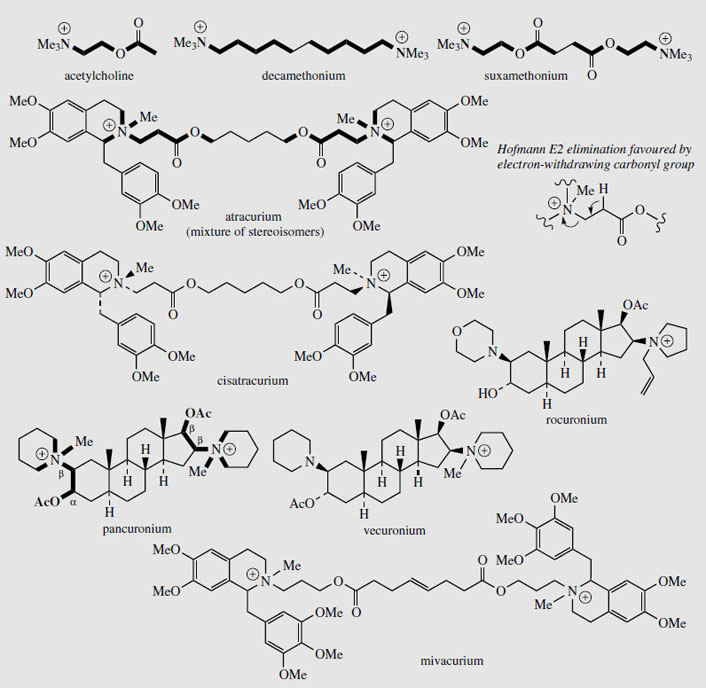 |
| Figure 49 |
Decamethonium (Figure 49) was the first synthetic curare-like muscle relaxant, but has since been superseded. In tubocurarine, the two nitrogens are also separated by ten atoms, and at physiological pHs it is likely that both centres will be positively charged. Obviously, the interatomic distance (1.4 nmin tubocurarine) is very dependent on the structure and stereochemistry rather than just the number of atoms separating the centres, but an extended conformation of decamethonium approximates to this distance. Suxamethonium (Figure 49) is an effective agent with a very short duration of action, due to the two ester functions, which are rapidly metabolized by an esterase (a pseudocholinesterase) in the body, and this means the period during which artificial respiration is required is considerably reduced. It also has ten-atom separation between the quaternary nitrogens. Atracurium (Figure 49) is a recent development, containing two quaternary nitrogens in benzyltetrahydroisoquinoline structures separated by 13 atoms. In addition to enzymic ester hydrolysis, atrocurium is also degraded in the body by non-enzymic E2 Hofmann elimination (Figure 49), which is independent of liver or kidney function. Normally, this elimination would require strongly alkaline conditions and a high temperature, but the presence of the carbonyl group increases the acidity and thus facilitates loss of the proton, and the elimination can proceed readily under physiological conditions, giving atracurium a half life of about 20 minutes. This is particularly valuable where patients have low or atypical pseudocholinesterase enzymes. Atracurium contains four chiral centres (including the quaternary nitrogens) and is supplied as a mixture of stereoisomers; the single isomer cisatracurium has now been introduced. This isomer is more potent than the mixture, has a slightly longer duration of action, and produces fewer cardiovascular side-effects. Mivacurium (Figure 49) has similar benzyltetrahydroisoquinoline structures to provide the quaternary centres, but the separation has now increased to 16 atoms. In pancuronium, separation of the two quaternary centres is achieved by a steroidal skeleton. This agent is about five times as potent as tubocurarine. Vecuronium is the equivalent monoquaternary structure, and has the fewest side-effects. Rocuronium is also based on a steroidal skeleton, and provides rapid action with no cardiovascular effects.
The toxiferines (Figure 85) also share the diquaternary character. Alcuronium is a semi-synthetic skeletal muscle relaxant containing the dimeric strychnine-like structure and is produced chemically from C-toxiferine.
These neuromuscular blocking agents act by occupying nicotinic acetylcholine (Figure 49) receptor sites. All the structures have two acetylcholine-like portions, which can interact with the receptor. Where these are built into a rigid framework, e.g. tubocurarine and pancuronium, the molecule probably spans and blocks several receptor sites. Tubocurarine and the heterocyclic analogues are termed non-depolarizing or competitive muscle relaxants. The straight chain structures, e.g. decamethonium and suxamethonium, initially mimic the action of acetylcholine but then persist at the receptor, and are termed depolarizing blocking agents. Thus they trigger a response, a brief contraction of the muscle, which is then followed by a prolonged period of muscular paralysis until the compound is metabolized.
Support our developers




